Lipofilicitet er muligvis den vigtigste fysisk-kemiske egenskab ved et potentielt lægemiddel; den spiller en rolle for opløselighed, absorption, membranpenetration, plasmaproteinbinding, distribution, CNS-penetration og fordeling i andre væv eller organer som f.eks. leveren og har en indvirkning på clearance-vejene. Det er vigtigt i forbindelse med ligandgenkendelse, ikke kun til målproteinet, men også CYP450-interaktioner, HERG-binding og PXR-medieret enzyminduktion.
LogP er en komponent i Lipinskis regel af 5 en tommelfingerregel til forudsigelse af opløselighed og permeabilitet, der er blevet et surrogat for lægemidlets lighed med andre lægemidler.
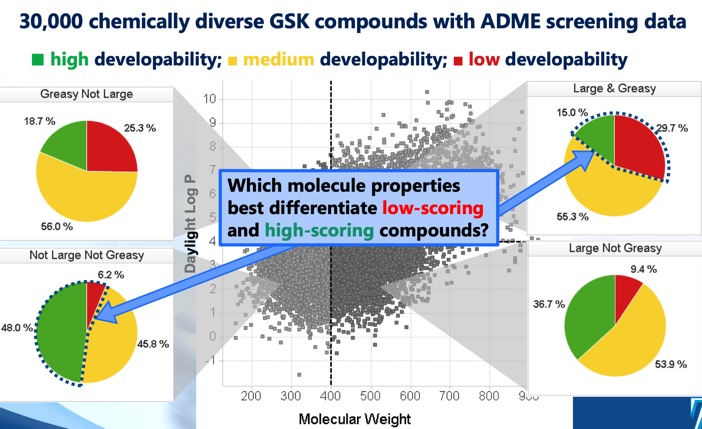
Den såkaldte Developability score DOI identificerer fire forskellige cLog P/molekylvægt-regioner, der definerer et optimalt og suboptimalt kemisk rum, og en udviklingsscore, der er afledt af regressionsmodeller ved hjælp af opløselighed, permeabilitet, proteinbinding og 3A4-hæmnings-screeningdata. Selv om sektoren MWt <400, cLogP <4 antydede den største chance for succes, blev det bemærket, at selv sektoren MWt >400, cLogP >4 omfattede nogle udviklelige molekyler, om end med en meget lavere chance for succes.
Det mest almindeligt anvendte mål for lipofilicitet er LogP, som er et molekyls fordelingskoefficient mellem en vandig og en lipofil fase, normalt oktanol og vand.
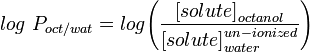
Måling af LogP kan foretages på en række forskellige måder, den mest almindelige er shake-flask-metoden, som består i at opløse noget af den pågældende opløste stof i et volumen octanol og vand, rystes i et tidsrum og derefter måles koncentrationen af den opløste stof i hvert opløsningsmiddel. Dette kan være tidskrævende, især hvis der ikke findes en hurtig spektroskopisk metode til at måle koncentrationen af molekylet i faserne. En hurtigere metode til log P-bestemmelse anvender højtydende væskekromatografi. Log P for en opløst stof kan bestemmes ved at korrelere dens retentionstid med lignende forbindelser med kendt log P-værdi doi.
Beregning af lipofilicitet
Sædvanligvis er det ikke praktisk muligt at bestemme log P for hver enkelt fremstillet forbindelse eksperimentelt (og det kan være af interesse at beregne log P forud for syntesen), og derfor anvendes beregnede resultater, der findes en række softwareværktøjer både desktop og online (må ikke anvendes til fortrolige forbindelser).
Mange af disse programmer fungerer ved hjælp af et stort træningsdatasæt med kendte værdier til bestemmelse af fragmentbidrag for understrukturer og funktionelle grupper, men logP er ikke en simpel additiv egenskab, og der er behov for korrektionstermer for at tage højde for nærhedseffekter, H-binding, elektroniske effekter osv. som vist i nedenstående eksempler.
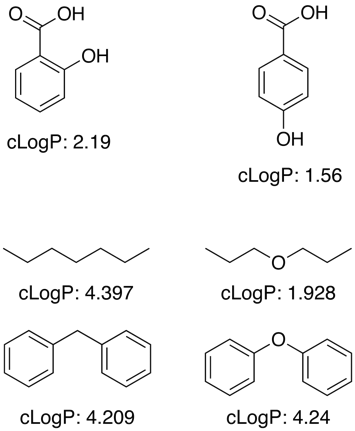
For ukendte funktionelle grupper foretager programmerne ofte en tilnærmelse ved hjælp af individuelle atombidrag.
De forskellige metoder til beregning af logP kan opdeles i tre forskellige fremgangsmåder.
Atomisk (f.eks. “AlogP”, ) & Forbedret atomisk / hybrid (“XlogP”, “SlogP”)
Fragment (“ClogP”, KlogP, ACD/logP)
Egenskabsbaserede metoder (“MlogP”, “VlogP”, “MClogP”, “TlogP”)
Atomisk logP tager hensyn til, at hvert atom har et bidrag til logP, og at bidragene til den endelige værdi er rent additive. Det er dog klart, at et nitrogen i et amid er anderledes end et nitrogen i en amin eller pyridin, Forbedret Atomic tager hensyn til atomtypen.
Fragmentmetoder anvender et stort træningsdatasæt med kendte værdier til at bestemme fragmentbidrag for understrukturer og funktionelle grupper, sammen med korrektionstermer til at tage hensyn til nærhedseffekter. Disse metoder falder ofte tilbage på atomare modeller for nye funktionelle grupper.
Egenskabsbaserede metoder har en tendens til at være beregningskrævende og egner sig ikke rigtig til at teste store datasæt.
Da træningssættene og algoritmerne varierer fra applikation til applikation, er det meget vigtigt ikke at kombinere beregnede resultater ved hjælp af forskellige værktøjer.
Nogle af værktøjerne giver brugeren mulighed for at udvide træningssættet ved hjælp af internt målte værdier, hvilket kan være kritisk, når man udforsker nye funktionelle grupper eller stilladser.
LogD
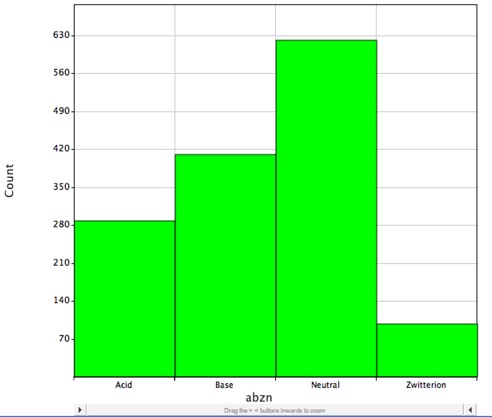
Men størstedelen af de kendte lægemidler indeholder ioniserbare grupper, som vist i nedenstående histogram, der viser fordelingen af små molekylære lægemidler med DrugBank og er sandsynligvis ladede ved fysiologisk pH, og LogP beskriver kun fordelingskoefficienten for neutrale (uladede) molekyler korrekt.
LogD fordelingskonstanten er en bedre deskriptor for et molekyls lipofilihed. Denne kan bestemmes på samme måde som LogP, men i stedet for at bruge vand justeres den vandige fase til en bestemt pH-værdi ved hjælp af en buffer. Log D er således pH-afhængig, og derfor skal man angive den pH, ved hvilken log D blev målt. Af særlig interesse er log D ved pH = 7,4 (den fysiologiske pH for blodserum).
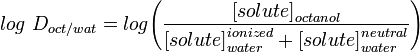
Anvendelser som Marvin giver brugeren mulighed for at beregne log D, men også vise pH-fordelingsprofilen, som vist nedenfor for Warfarin.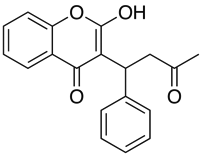
For forbindelser med en pKa tæt på den fysiologiske pH kan det være kritisk at overveje, hvad der rent faktisk kan være den fremherskende ioniserede form.
Dette kan også være værdifuldt, når man tænker på absorption fra de forskellige regioner i fordøjelseskanalen, hvor pH-værdien varierer fra 1-3 i maven til 7-8 i ileum.
De forskellige funktionelle gruppers bidrag til LogD er blevet undersøgt “LogD contributions of substituents commonly used in medicinal chemistry” DOI, denne undersøgelse anvendte matched molecular pairs analyse af eksperimentelle LogD-værdier fra flere tusinde forbindelser indsamlet ved hjælp af shake-flask-metoden ved pH = 7,4. De rapporterede den gennemsnitlige deltaLogD-difference for bestemte molekylpar, og resultaterne er vist nedenfor for det tilfælde, hvor den funktionelle gruppe befinder sig på en hvilken som helst position på phenylringen. Jeg har også inkluderet den beregnede LogD ved hjælp af Chemaxon-software.
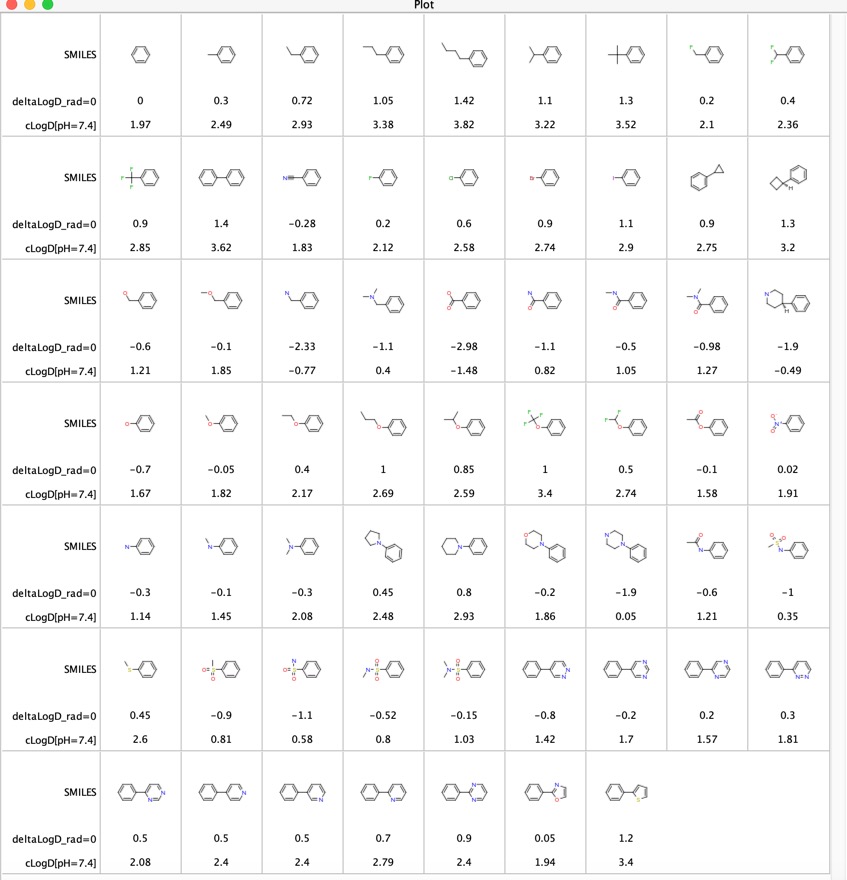
Dette er en nyttig tabel til sammenligning af funktionelle grupper, især de sidste 11 poster sammenligner den indflydelse, som forskellige heterocyklers indflydelse har på LogD. Disse heterocyklusser anvendes ofte som bioisosteriske erstatninger for en phenylring.
Jeg tænkte, at det kunne være interessant at sammenligne LogD-forskellene bestemt ved hjælp af de matchede molekylepar (deltaLogD_rad=0) med de værdier, der er bestemt ved hjælp af Chemaxon-beregnet LogD (deltacLogD). Som du kan se nedenfor er der en ret god overensstemmelse.
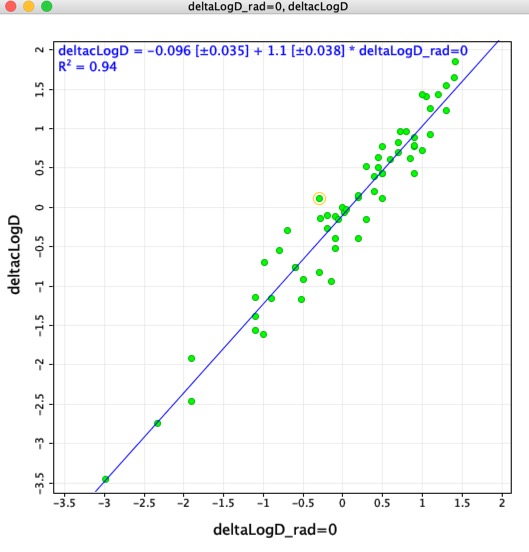
Husk Da træningssættene og algoritmerne varierer fra program til program, er det meget vigtigt ikke at kombinere beregnede resultater ved hjælp af forskellige værktøjer.
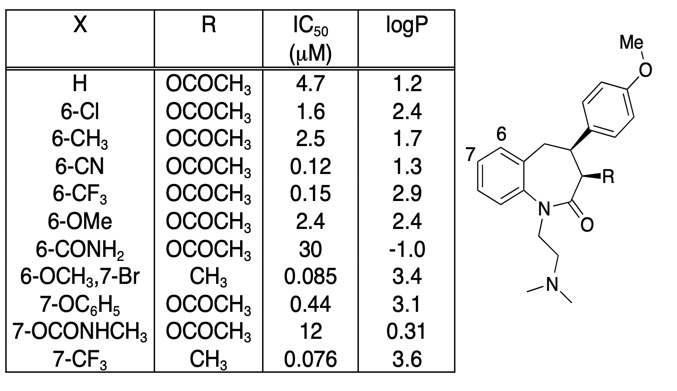
Det er vigtigt at være opmærksom på, at enhver forbedring i bindingsaffinitet ikke udelukkende er drevet af en stigning i LogD, det er ofte nyttigt blot at plotte bindingsaffinitet versus LogD. De mere interessante sammensætningsmodifikationer er ikke nødvendigvis dem, der giver den største stigning i affiniteten, men kan være dem, der giver en stigning i affiniteten uden en tilsvarende stigning i lipofiliciteten. Hvis man ser på nedenstående tabel, er der en række forbindelser med meget høj affinitet.
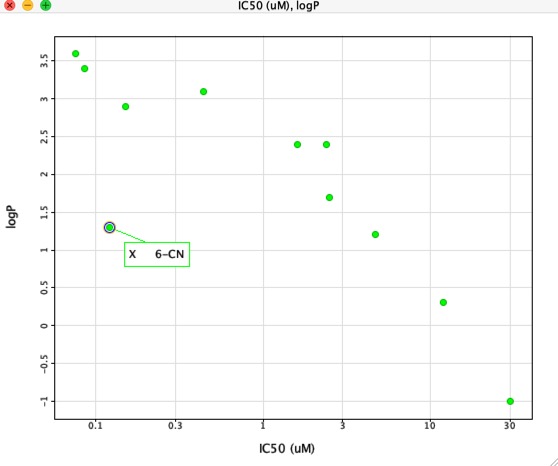
Hvis man imidlertid plotter IC50 over for LogP som vist nedenfor, er der en meget klar sammenhæng mellem LogP og IC50, men én forbindelse er dog klart anderledes. 6-CN-substituenten giver en stigning i affinitet uden en tilsvarende stigning i LogP.
Lipofilicitet er også en vigtig komponent, og mange af off-target-ansvarlighederne, herunder plasmaproteinbinding (især albumin), HERG, CYP-interaktioner og transportere, har stærke korrelationer med lipofilicitet, og der har været en række undersøgelser, der kæder et højt logP sammen med sandsynligheden for, at stoffer mislykkes under udviklingen som følge af dårlige ADMET-egenskaber (absorption, distribution, metabolisme, udskillelse og toksicitet). Derimod er det ofte klart, at der kræves en vis størrelse og lipofilicitet for at opnå rimelige affinitetsniveauer. At balancere disse krav er en central udfordring i forbindelse med lægemiddeludvikling, og det foreslås, at kemikere sigter mod det søde punkt, MWt 250-500 og LogP 2-4. En konsekvens af denne fremgangsmåde er, at det er nødvendigt at prioritere lavmolekylære, mindre lipofile forbindelser fra screening. Medicinsk kemis indledende fokus bør være at udvælge udgangspunkter af god kvalitet og derefter at kontrollere forskydninger i fysisk-kemiske egenskaber effektivt under optimeringsprocessen.
Værd at læse
Finding the sweet spot: the role of nature and nurture in medicinal chemistry DOI
Lipofilic efficiency: the most important efficiency metric in medicinal chemistry DOI
Sidst opdateret 12. januar 2019