Morbillivirusser er meget smitsomme patogener og er ansvarlige for forskellige udbrud i uudsatte befolkninger (Pfeffermann et al., 2018). De tilhører ordenen Mononegavirales og familien Paramyxoviridae og er karakteriseret ved et ikke-segmenteret, lineært, lineært, negativt-strenget RNA-genom (Lamb og Parks, 2013). Morbillivirus er kendetegnet ved at forårsage moderat til alvorlig respiratorisk, gastrointestinal, immunosuppression og/eller neurologiske sygdomme hos en lang række værter, herunder mennesker (mæslingevirus), kødædende dyr (hunde-morbillivirus, tidligere hundesygevirus), kvæg (rinderpestvirus), delfiner og marsvin og andre vildtlevende truede arter (Lamb og Parks, 2013; Martinez-Gutierrez og Ruiz-Saenz, 2016).
Measlesvirus (MeV) og canine morbillivirus (CDV) anses for at være de mest smitsomme virus blandt denne familie (De Vries et al, 2015), og på grund af CDV’s høje transmissionspotentiale samt dets transmissionspotentiale på tværs af arter er de globale sundheds- og naturbeskyttelsesmyndigheder meget bekymrede over CDV’s rolle for bevarelsen af truede arter og det mulige “spring” fra dyr til mennesker (Terio og Craft, 2013; Ohishi et al., 2014). Tamme hunde er den vigtigste vært for CDV og kunne også betragtes som et reservoir for andre pattedyr (Suzuki et al., 2015; Duque-valencia et al., 2019); men baseret på CDV’s biologi kunne mennesker også blive et potentielt mål (Cosby og Weir, 2018; Rendon-Marin et al, 2019).
For at forsøge at forstå den potentielle risiko for overførsel af CDV til mennesker er det nødvendigt at indsamle alle eksisterende beviser; og undersøgelsen af dette agens oprindelse og spredning i hundepopulationen kunne udgøre en vigtig nøgle til at forstå denne proces. For nylig blev der offentliggjort en artikel i International Journal of Paleopathology, som indbød til en diskussion om CDV’s evolutionære oprindelse. Det konkluderes, at CDV opstod som et pandemisk patogen i Sydamerika efter infektion og tilpasning af MeV til hunde i løbet af den sydamerikanske koloniseringsperiode. Dette resultat blev opnået via en tværfaglig tilgang, der blev vedtaget ved at syntetisere en palæopatologisk analyse af 96 præcolumbianske hunde (750-1470 CE) fra Weyanoke Old Town-området i Virginia, med historiske rapporter, molekylær analyse og morbilliviral epidemiologi (Uhl et al, 2019).
Noteringsmæssigt forsvandt indfødte hundepopulationer fra Amerika næsten efter koloniseringsperioden, og europæiske og eurasiske hunde blev introduceret til kontinentet, hvilket efterlod kun lidt genetisk baggrund af dens amerikanske forgængere (Ni Leathlobhair et al., 2018). En anden vigtig faktor, der er værd at overveje, er, at “ukendte” sygdomme også kan være blevet introduceret, hvilket gør det sværere at spore oprindelsen af nye patogener. Desuden kunne et kunstigt selektionstryk over for tamme hunde og endda menneskelige populationer, især i koloniseringsperioden, have øget sygdomsforekomsten og dermed begrænset den genetiske variation (Ostrander et al, 2017), hvilket igen kunne betyde en mindre effektiv reaktion mod patogener.
Men blandt disse “nye” patogener/sygdomme blev CDV først beskrevet af Antonio de Ulloa y de la Torre-Giral i 1746 som en sygdom, der ramte hunde i Quito-regionen og de andre dele af Sydamerika, og den blev kort efter rapporteret i Europa. CDV blev registreret i Spanien i 1760 med 900 dødsfald på en enkelt dag i Madrid, og tre år senere, dvs. i 1764 og 1770, var den nået til henholdsvis Storbritannien og Italien (Blancou, 2004). Virusoverførbarhed og større modtagelighed hos hvalpe sammenlignet med voksne hunde blev senere rapporteret af Edward Jenner i begyndelsen af 1800-tallet. Han sammenlignede deres overførbarhed med MeV og opdagede, at overlevende var beskyttet mod efterfølgende infektion (Jenner, 1809; Nambulli et al., 2016).
Kort sagt, efter ankomsten af europæiske pionerer i det femtende århundrede blev nye smitsomme sygdomme velsagtens den mest ødelæggende konsekvens af koloniseringen, fordi de oprindelige amerikanske befolkninger ikke tidligere havde været udsat for patogener, der var blevet almindelige i Europa (Walker et al., 2015). Flere mæslingeepidemier ødelagde derfor de oprindelige amerikanske befolkninger (Walker et al., 2015; Nambulli et al., 2016). Uhl et al. via en blandet tilgang af palæopatologiske, historiske, molekylære og epidemiologiske beviser rapporterede, at alvorlige MeV-epidemier i de oprindelige amerikanske befolkninger lettede springet af MeV til store tamme hundepopulationer i bymiljøer i Sydamerika og tilpasningen af viruset som endemisk CDV (Uhl et al., 2019). Historiske optegnelser kunne også bevise, at få år efter denne tilpasning til sydamerikanske hunde blev CDV transporteret til Europa i 1760, hvor det i første omgang fremkaldte udbredte epidemier med høj dødelighed, før det blev endemisk (Jenner, 1809).
Derimod blev molekylær fylogeografi i forbindelse med evolutionære forudsigelser og tiden til den seneste fælles forfader (tMRCA) beregnet for CDV’s oprindelse i USA i 1880’erne (95% højeste posterior tæthed, 1858-1913) (Panzera et al, 2015), hvilket klart modsiger beskrivelsen af viruset i Europa i det attende århundrede. Sekvensanalyser, der førte til denne hypotese, skal undersøges omhyggeligt på grund af bias og den begrænsede tilgængelighed af sekvenser, der blev brugt i denne molekylære fylogeografiske rekonstruktion. Desuden er mange af de oprindelige forfædres sekvenser gået tabt på grund af CDV’s og andre morbillivirers virale RNA-genomers labilitet. Disse faktorer har givet anledning til at sætte spørgsmålstegn ved nytten af de nuværende tMRCA-beregninger for RNA-virus (Sharp og Simmonds, 2011; Nambulli et al., 2016).
Ifølge Uhl et al, morbillivirus kan være opstået fra kvæg omkring 376 f.Kr. på det “gamle kontinent” (figur 1), og domesticering af dyr kan have haft en betydelig indflydelse på begivenheder på tværs af arter, hvilket sandsynligvis sporer et startpunkt for MeV-fremkomsten til ca. 900 AC (Uhl et al., 2019). I modsætning til de nuværende CDV-fylogenetiske rekonstruktioner understøttes MeV-divergensen stærkt af den afslappede clock Bayesian-fylogenetiske analyse. Divergenstidspunktet mellem MeV og rinderpestvirus var blevet vist at have fundet sted i ca. det ellevte til tolvte århundrede (Furuse et al., 2010). Andre molekylære data, såsom tilstedeværelsen af et nyt morbillivirus (nært beslægtet med CDV og PDV), der cirkulerer i flagermus fra Brasilien (DrMV), giver mulighed for at spekulere i, at CDV og DrMV måske har en fælles sydamerikansk forfader (Drexler et al., 2012) og dermed indirekte støtter ideen om CDV’s tidlige sydamerikanske oprindelse.
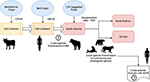
Figur 1. Skematisk fremstilling af den mulige evolutionære transmissionsvej for canin morbillivirus (CDV). Se teksten for referencer.
Ud over den epistemologiske og/eller videnskabelige betydning af den geografiske oprindelse og datoen for CDV-divergens er der vigtige spor, som skal afklares for bedre at forstå CDV’ernes nuværende indvirkning på overførsel mellem arter, dyrebevaring og zoonotisk potentiale (figur 1). Det er klart, at i modsætning til MeV-infektionen, som opretholdes af en enkelt vært (mennesker), er CDV blevet bredt bevist at være et promiskuøst patogen, der forårsager infektion/sygdom i en lang række kødædende og ikke-kødædende arter (Martinez-Gutierrez og Ruiz-Saenz, 2016). Denne promiskuitet er blevet tilskrevet ikke kun CDV-hæmagglutininets (H) evne til at interagere med værtscellereceptorer, såsom SLAM i mononukleære celler og nectin-4 i epitelceller, men også ligheden mellem arternes sekvenser af de ovennævnte receptorer (Rendon-Marin et al., 2019). Aminosyreligheden blandt pattedyrs SLAM-receptorer, herunder havpattedyr, er >80% (Ohishi et al., 2014), hvilket understøtter resultaterne af overførsel på tværs af arter. Derudover er der en mangel på artsrelateret variation i nectin-4-sekvenserne blandt mennesker, mus og hunde, fordi menneskelig nectin-4 kunne fungere som en in vitro-receptor for CDV (Noyce et al., 2011).
Naturlige CDV-udbrud hos forskellige ikke-menneskelige primater har givet anledning til bekymring med hensyn til mulig overførsel af CDV til mennesker (Yoshikawa et al., 1989; Sun et al., 2010; Qiu et al., 2011; Sakai et al., 2013a). Der er rapporter om, at CDV-stammer fra aber har den iboende evne til at bruge humant nectin-4 til virusindgang, og at disse CDV-stammer fra aber let tilpasser sig til at bruge den humane CD150-receptor (SLAM) efter minimale aminosyreændringer i det virale H-protein (Bieringer et al., 2013; Sakai et al., 2013b). På grundlag af den eksperimentelle in vivo CDV-infektion af Cynomolgus-makaker (Macaca fascicularis) i tilstedeværelse af MeV-immunitet var makakerne imidlertid delvist krydsbeskyttet mod CDV-udfordringen (De Vries et al., 2014). Dette tyder på, at selv om CDV let kan inficere primater, er MeV-immunitet beskyttende, og at CDV-infektion kan være selvbegrænsende. Hvis man overfører dette resultat til mennesker, er der en potentiel risiko for CDV-infektion hos mennesker, der mangler tværbeskyttende MeV-immunitet på grund af manglende vaccination og vaccinefejl (Haralambieva et al., 2015) eller på grund af manglende vaccination i den mulige post-udryddelsesæra (Holzmann et al., 2016).
“Emerging viruses” kunne angiveligt opstå via overførsel af virus fra dyr til mennesker på tværs af arter (Wolfe et al., 2007). Nye undersøgelser, både strukturelle og bioinformatiske, tyder på, at blot en enkelt aminosyreændring i en proteinsekvens kan være nok til at overvinde begrænsningen i brugen af cellulære receptorer blandt to forskellige værter, f.eks. mennesker og drøvtyggere (Abdullah et al., 2018). En unik mutation i CDV H-proteinet in vitro gør det muligt for dette patogen at inficere celler, der udtrykker den menneskelige SLAM-receptor (Otsuki et al., 2013). Hvis vi omfavner hypotesen om, at CDV udviklede sig fra MeV, kunne det desuden være muligt, at en CDV-afstamning kunne være i stand til at re-inficere mennesker på grund af den kontinuerlige udvikling af både virus og mennesker, som det tidligere er blevet foreslået i andre modeller, selv om den forfædte “jumpervirus” var forsvundet fra jorden for længe siden (Emerman og Malik, 2010).
Dertil kommer, at et af de mest interessante resultater præsenteret af Uhl et al. er optimeringen af både CDV- og MeV-generne til menneskelig codon use bias (CUB), hvilket tyder på, at CDV’s codonbrug er tættere på menneskelig CUB end hunde-CUB, fordi virussen eller dens stamfader, højst sandsynligt MeV, oprindeligt blev tilpasset mennesker (Uhl et al, 2019). CUB henviser til det fænomen, hvor nogle synonyme kodoner anvendes oftere end andre, og hvordan denne præference varierer inden for og mellem arter (Behura og Severson, 2013). I RNA-virus er kodonanvendelsen under udvælgelse, fordi virusene er fuldstændig afhængige af værtens tRNA’er, og skævheden skyldes, at virusene matcher deres værts codonanvendelse (Jenkins og Holmes, 2003). Evolutionen kan undertiden favorisere virus, der matcher deres værts kodonbrug for at fremme replikationshastigheden og tilpasningen til værten, som det er blevet rapporteret i andre RNA-virus (Goni et al., 2012; Lauring et al., 2012; Di Paola et al., 2018; Freire et al., 2018).
Sidst vil vi gerne argumentere for, at nogle andre faktorer skal tages i betragtning i det mulige zoonotiske scenarie for CDV. Krydsneutralisering mellem MeV og CDV har været anerkendt i mange år (Brown og Mccarthy, 1974), og denne forudsætning har eksisteret i mere end et halvt århundrede, da MeV-vaccinen blev brugt til at beskytte hvalpe mod CDV i en alder, hvor passiv moderlig immunitet ofte forstyrrede CDV-vaccinationen (Baker et al., 1966; Brown et al., 1972). Ikke desto mindre anbefales det stadig at anvende en kommerciel dobbelt CDV/MeV-vaccine til vaccination i tilstedeværelse af moderens immunitet, og vaccinen har været nyttig mod klinisk mæslingesygdom hos ikke-menneskelige primater (Christe et al., 2019). Derfor kan man spekulere i, at MeV-flokimmunitet undgår CDV-hop og mulig gentilpasning til mennesker via overførsel gennem hunde eller dyr i naturen.
Sluttende bemærkninger
Evolution og oprindelse af virale patogener kan ikke let studeres; herefter er en tværfaglig tilgang nødvendig for at forstå og måske forudsige nye mulige virale trusler mod mennesker. På grund af deres særlige biologi udgør virale patogener som CDV en unik model til at forstå spring mellem arter og zoonotisk potentiale af virale agenser meget tæt på den menneskelige befolkning. Ud over de traditionelle molekylærfylogenetiske undersøgelser og palæopatologiske arbejder må forskerne anvende forskellige metoder til at undersøge CDV’s oprindelse og de nuværende virale krav og værtsbetingelser for at springe mellem arter. Indførelsen af beregningsmetoder, såsom strukturel bioinformatik og palæovirologiske undersøgelser, kunne bidrage til forudsigelse og forebyggelse eller i det mindste give en bedre forståelse af denne nye og måske zoonotiske sygdom fra et andet perspektiv, hvor ikke kun sekventeringsdata, men også strukturer og funktioner betragtes som nøgleoplysninger til dette formål.
Author Contributions
Alle forfattere på listen har ydet et væsentligt, direkte og intellektuelt bidrag til arbejdet og godkendt det med henblik på offentliggørelse.
Funding
Dette arbejde blev finansielt støttet af Departamento Administrativo de Ciencia, Tecnología e Innovación-COLCIENCIAS Grant No. 123171249669 til JR-S.
Interessekonflikter
Forfatterne erklærer, at forskningen blev udført uden kommercielle eller finansielle forbindelser, der kunne opfattes som en potentiel interessekonflikt.
Brown, A. L., og Mccarthy, R. E. (1974). Forholdet mellem mæslinge- og hundesygevirus bestemt ved forsinkede overfølsomhedsreaktioner af forsinket type hos hunde. Nature 248, 344-345. doi: 10.1038/248344a0
PubMed Abstract | CrossRef Full Text | Google Scholar