Morbillivirusy są wysoce zakaźnymi patogenami i są odpowiedzialne za różne ogniska w nieeksponowanych populacjach (Pfeffermann i in., 2018). Należą do rzędu Mononegavirales oraz rodziny Paramyxoviridae i charakteryzują się niesegmentowanym, liniowym, ujemnie prążkowanym genomem RNA (Lamb i Parks, 2013). Morbillivirusy są wyróżniane ze względu na wywoływanie umiarkowanych do ciężkich chorób układu oddechowego, pokarmowego, immunosupresji i/lub chorób neurologicznych u szerokiej gamy żywicieli, w tym ludzi (wirus odry), mięsożerców (canine morbillivirus dawniej canine distemper virus), bydła (wirus księgosuszu), delfinów i morświnów oraz innych gatunków zagrożonych dzikim życiem (Lamb i Parks, 2013; Martinez-Gutierrez i Ruiz-Saenz, 2016).
Wirusy Measles (MeV) i canine morbillivirus (CDV) są uważane za najbardziej zaraźliwe wirusy wśród tej rodziny (De Vries i in., 2015), a ze względu na wysoki potencjał transmisyjny CDV, jak również możliwość jego przenoszenia międzygatunkowego, światowe władze zajmujące się zdrowiem oraz ochroną przyrody są bardzo zaniepokojone rolą CDV w ochronie gatunków zagrożonych wyginięciem oraz możliwym „przeskokiem” ze zwierząt na ludzi (Terio i Craft, 2013; Ohishi i in., 2014). Psy domowe są głównym gospodarzem dla CDV i mogą być również uważane za rezerwuar dla innych ssaków (Suzuki i in., 2015; Duque-valencia i in., 2019); jednak w oparciu o biologię CDV, ludzie mogą również stać się potencjalnym celem (Cosby i Weir, 2018; Rendon-Marin i in., 2019).
Próbując zrozumieć potencjalne ryzyko przeniesienia CDV na ludzi, konieczne jest zebranie wszystkich istniejących dowodów; a badanie pochodzenia i rozprzestrzeniania się tego czynnika w populacji psów może stanowić ważny klucz do zrozumienia tego procesu. Ostatnio, praca opublikowana w International Journal of Paleopathology zachęciła do dyskusji na temat ewolucyjnego pochodzenia wirusa CDV. Stwierdzono w niej, że CDV powstał jako patogen pandemiczny w Ameryce Południowej w następstwie zakażenia i adaptacji MeV u psów w okresie kolonizacji Ameryki Południowej. Wynik ten uzyskano dzięki interdyscyplinarnemu podejściu przyjętemu poprzez syntezę analizy paleopatologicznej 96 prekolumbijskich psów (750-1470 CE) ze stanowiska Weyanoke Old Town, Virginia, z doniesieniami historycznymi, analizą molekularną i epidemiologią morbilliwirusową (Uhl i in., 2019).
Na uwagę zasługuje fakt, że rodzime populacje psów z Ameryki niemal zanikły po okresie kolonizacji, a na kontynent wprowadzono psy europejskie i euroazjatyckie, pozostawiając niewielkie zaplecze genetyczne swoich amerykańskich poprzedników (Ni Leathlobhair i in., 2018). Innym ważnym czynnikiem wartym rozważenia jest fakt, że mogły zostać wprowadzone również „nieznane” choroby, co utrudnia śledzenie pochodzenia nowych patogenów. Co więcej, sztuczna presja selekcyjna nad psami domowymi, a nawet populacjami ludzkimi, szczególnie w okresie kolonizacji, mogła zwiększyć częstość występowania chorób, ograniczając tym samym zmienność genetyczną (Ostrander i in., 2017), co z kolei mogło oznaczać mniej skuteczną odpowiedź przeciwko patogenom.
Pośród tych „nowych” patogenów/chorób, CDV został po raz pierwszy opisany przez Antonio de Ulloa y de la Torre-Giral w 1746 roku jako choroba dotykająca psy w regionie Quito i innych częściach Ameryki Południowej, a wkrótce potem został zgłoszony w Europie. W Hiszpanii CDV został odnotowany w 1760 r., kiedy to w ciągu jednego dnia w Madrycie doszło do 900 zgonów, a 3 lata później, tj. w 1764 i 1770 r. dotarł odpowiednio do Wielkiej Brytanii i Włoch (Blancou, 2004). O przenoszeniu się wirusa i większej podatności szczeniąt w porównaniu z dorosłymi psami donosił później Edward Jenner na początku XIX wieku. Porównał ich transmisyjność z tą u MeV i odkrył, że te, które przeżyły, były chronione przed późniejszą infekcją (Jenner, 1809; Nambulli et al., 2016).
W skrócie, po przybyciu europejskich pionierów w XV wieku, nowe choroby zakaźne prawdopodobnie stały się najbardziej niszczycielską konsekwencją kolonizacji, ponieważ rdzenne populacje amerykańskie nie miały wcześniejszej ekspozycji na patogeny, które stały się powszechne w Europie (Walker et al., 2015). Wielokrotne epidemie odry wyniszczały zatem rdzenne populacje amerykańskie (Walker i in., 2015; Nambulli i in., 2016). Uhl i wsp. poprzez mieszane podejście do dowodów paleopatologicznych, historycznych, molekularnych i epidemiologicznych, podali, że ciężkie epidemie MeV w rdzennych populacjach amerykańskich ułatwiły przeskok MeV do dużych populacji psów domowych w środowiskach miejskich w Ameryce Południowej i adaptację wirusa jako endemicznego CDV (Uhl i wsp., 2019). Również zapisy historyczne mogą dowodzić, że kilka lat po tej adaptacji do południowoamerykańskich psów, CDV został przetransportowany do Europy w 1760 roku, gdzie początkowo wywołał szeroko rozpowszechnione epidemie z wysoką śmiertelnością, zanim stał się endemiczny (Jenner, 1809).
Jednakże filogeografia molekularna związana z przewidywaniami ewolucyjnymi oraz czas do najnowszego wspólnego przodka (tMRCA) zostały obliczone dla pochodzenia CDV w Stanach Zjednoczonych w latach osiemdziesiątych XIX wieku (95% najwyższa gęstość potomna, 1858-1913) (Panzera i in., 2015), co jest wyraźnie sprzeczne z opisem wirusa w Europie w XVIII wieku. Analizy sekwencji, które doprowadziły do tej hipotezy, muszą być dokładnie zbadane ze względu na stronniczość i ograniczoną dostępność sekwencji, które zostały wykorzystane w tej rekonstrukcji filogeografii molekularnej. Ponadto, wiele oryginalnych sekwencji przodków zostało utraconych z powodu niestabilności genomu RNA wirusa CDV i innych wirusów morbillivirusa. Czynniki te dały podstawę do zakwestionowania użyteczności obecnych obliczeń tMRCA dla wirusów RNA (Sharp i Simmonds, 2011; Nambulli i in., 2016).
Według Uhl i in., morbillivirus mógł pochodzić od bydła około 376 p.n.e. na „starym kontynencie” (rysunek 1), a udomowienie zwierząt mogło mieć znaczący wpływ na wydarzenia międzygatunkowe, prawdopodobnie śledząc punkt początkowy w pojawieniu się MeV do około 900 AC (Uhl i in., 2019). W przeciwieństwie do obecnych rekonstrukcji filogenetycznych CDV, dywergencja MeV jest silnie wspierana przez analizę filogenetyczną Bayesian relaxed clock. Wykazano, że czas dywergencji między MeV a wirusem księgosuszu miał miejsce w przybliżeniu w XI-XII wieku (Furuse i in., 2010). Inne dane molekularne, takie jak obecność nowego wirusa morbillivirus (blisko spokrewnionego z CDV i PDV) krążącego wśród nietoperzy z Brazylii (DrMV), pozwalają spekulować, że CDV i DrMV mogą mieć wspólnego południowoamerykańskiego przodka (Drexler i in., 2012), tym samym pośrednio wspierając koncepcję wczesnego południowoamerykańskiego pochodzenia CDV.
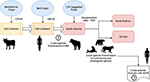
Rysunek 1. Schematyczne przedstawienie możliwej ewolucyjnej drogi przenoszenia morbillivirusa psów (CDV). Patrz tekst w celu uzyskania odniesień.
Poza epistemologicznym i/lub naukowym znaczeniem pochodzenia geograficznego i daty dywergencji CDV, istnieją ważne wskazówki, które muszą zostać wyjaśnione, aby lepiej zrozumieć obecny wpływ CDV na transmisję międzygatunkową, ochronę zwierząt i potencjał zoonotyczny (rysunek 1). Oczywiste jest, że w przeciwieństwie do zakażenia MeV, które jest podtrzymywane przez jednego żywiciela (człowieka), CDV okazał się być patogenem promiskuitycznym, wywołującym zakażenie/chorobę u szerokiej gamy gatunków mięsożernych i nie mięsożernych (Martinez-Gutierrez i Ruiz-Saenz, 2016). Ta promiskuityczność została przypisana nie tylko zdolności hemaglutyniny (H) CDV do interakcji z receptorami komórkowymi gospodarza, takimi jak SLAM w komórkach jednojądrzastych i nektyna-4 w komórkach nabłonkowych, ale także podobieństwu między gatunkami sekwencji wspomnianych receptorów (Rendon-Marin i in., 2019). Podobieństwo aminokwasów wśród receptorów SLAM ssaków, w tym ssaków morskich, wynosi >80% (Ohishi i in., 2014), potwierdzając tym samym wyniki transmisji międzygatunkowej. Ponadto brak jest gatunkowej zmienności w sekwencjach nektyny-4 wśród ludzi, myszy i psów, ponieważ ludzka nektyna-4 może funkcjonować jako receptor in vitro dla CDV (Noyce i in., 2011).
Naturalne ogniska CDV u różnych naczelnych wzbudziły obawy dotyczące możliwej transmisji CDV na ludzi (Yoshikawa i in., 1989; Sun i in., 2010; Qiu i in., 2011; Sakai i in., 2013a). Istnieją doniesienia, że małpie szczepy CDV mają wewnętrzną zdolność do wykorzystywania ludzkiej nektyny-4 do wnikania wirusa oraz że te małpie CDV łatwo adaptują się do wykorzystywania ludzkiego receptora CD150 (SLAM) po minimalnych zmianach aminokwasowych w wirusowym białku H (Bieringer i in., 2013; Sakai i in., 2013b). Jednakże, w oparciu o eksperymentalne zakażenie in vivo CDV makaków Cynomolgus (Macaca fascicularis) w obecności odporności MeV, makaki były częściowo chronione krzyżowo przed wyzwaniem CDV (De Vries et al., 2014). Sugeruje to, że chociaż CDV może łatwo zainfekować naczelne, odporność MeV jest ochronna i że infekcja CDV może być samoograniczająca. Przenosząc ten wynik na ludzi, istnieje potencjalne ryzyko zakażenia CDV u osób, którym brakuje krzyżowej odporności ochronnej MeV z powodu nieszczepienia i niepowodzenia szczepień (Haralambieva i in., 2015) lub z powodu braku szczepień w możliwej erze po eradykacji (Holzmann i in., 2016).
„Pojawiające się wirusy” mogą podobno powstawać poprzez międzygatunkowe przenoszenie wirusów ze zwierząt na ludzi (Wolfe i in., 2007). Nowe badania, zarówno strukturalne, jak i bioinformatyczne, sugerują, że tylko jedna zmiana aminokwasowa w sekwencji białka może wystarczyć do pokonania ograniczenia w wykorzystaniu receptorów komórkowych między dwoma różnymi gospodarzami, takimi jak ludzie i przeżuwacze (Abdullah i in., 2018). Unikalna mutacja w białku H CDV in vitro umożliwia temu patogenowi infekowanie komórek wyrażających ludzki receptor SLAM (Otsuki i in., 2013). Ponadto, jeśli przyjmiemy hipotezę, że CDV wyewoluował z MeV, może być możliwe, że potomek CDV będzie w stanie ponownie zainfekować ludzi z powodu ciągłej ewolucji zarówno wirusa, jak i ludzi, co zostało wcześniej zasugerowane w innych modelach, nawet jeśli przodek „wirusa skoczka” zniknął z Ziemi jakiś czas temu (Emerman i Malik, 2010).
Ponadto, jednym z najciekawszych wyników przedstawionych przez Uhl i wsp. jest optymalizacja zarówno genów CDV, jak i MeV pod kątem ludzkiej tendencyjności użycia kodonów (CUB), sugerując, że użycie kodonów CDV jest bliższe ludzkiej CUB niż psiej CUB, ponieważ wirus lub jego protoplasta, najprawdopodobniej MeV, został początkowo dostosowany do ludzi (Uhl i wsp., 2019). CUB odnosi się do zjawiska, w którym niektóre synonimiczne kodony są używane częściej niż inne i jak ta preferencja różni się w obrębie i wśród gatunków (Behura i Severson, 2013). W wirusach RNA użycie kodonów podlega selekcji, ponieważ wirusy są całkowicie zależne od tRNA gospodarza, a tendencyjność wynika z dopasowania wirusów do użycia kodonów ich gospodarzy (Jenkins i Holmes, 2003). Ewolucja może czasami sprzyjać wirusom, które pasują do wykorzystania kodonów gospodarza, aby promować szybkość replikacji i adaptację do gospodarza, jak zgłoszono w innych wirusach RNA (Goni i in., 2012; Lauring i in., 2012; Di Paola i in., 2018; Freire i in., 2018).
Na koniec chcielibyśmy argumentować, że niektóre inne czynniki muszą być brane pod uwagę w możliwym scenariuszu zoonotycznym CDV. Neutralizacja krzyżowa między MeV i CDV została uznana od wielu lat (Brown i Mccarthy, 1974), a przesłanka ta istniała przez ponad pół wieku, kiedy szczepionka MeV była używana do ochrony szczeniąt przed CDV w wieku, w którym bierna odporność matki często kolidowała ze szczepieniem CDV (Baker i in., 1966; Brown i in., 1972). Mimo to stosowanie komercyjnej podwójnej szczepionki CDV/MeV jest nadal zalecane do szczepień w obecności odporności matczynej, a szczepionka ta okazała się przydatna w zwalczaniu klinicznej choroby odry u ssaków naczelnych (Christe i in., 2019). Stąd można spekulować, że odporność stadna MeV pozwala uniknąć przeskoku CDV i możliwej readaptacji do ludzi poprzez transmisję przez psy lub dzikie zwierzęta.
Uwagi końcowe
Ewolucja i pochodzenie patogenów wirusowych nie mogą być łatwo zbadane; w związku z tym konieczne jest podejście multidyscyplinarne, aby zrozumieć i być może przewidzieć nowe możliwe zagrożenia wirusowe dla ludzi. Ze względu na ich osobliwą biologię, wirusowe patogeny takie jak CDV stanowią unikalny model dla zrozumienia międzygatunkowego przemieszczania się i potencjału zoonotycznego czynników wirusowych bardzo blisko populacji ludzkiej. Poza tradycyjnymi molekularnymi badaniami filogenetycznymi i pracami paleopatologicznymi, naukowcy muszą przyjąć różne podejścia, aby zbadać pochodzenie CDV i obecne wymagania wirusa i gospodarza dla skoków międzygatunkowych. Wprowadzenie metod obliczeniowych, takich jak bioinformatyka strukturalna i badania paleowirologiczne, mogłoby pomóc w przewidywaniu i zapobieganiu lub przynajmniej zapewnić lepsze zrozumienie tej wyłaniającej się i być może odzwierzęcej choroby z innej perspektywy, biorąc pod uwagę nie tylko dane sekwencjonowania, ale także struktury i funkcje jako kluczowe informacje do tego celu.
Wkład autorów
Wszyscy wymienieni autorzy wnieśli istotny, bezpośredni i intelektualny wkład w tę pracę i zatwierdzili ją do publikacji.
Funding
Ta praca została wsparta finansowo przez Departamento Administrativo de Ciencia, Tecnología e Innovación-COLCIENCIAS Grant No. 123171249669 dla JR-S.
Oświadczenie o konflikcie interesów
Autorzy oświadczają, że badania zostały przeprowadzone przy braku jakichkolwiek komercyjnych lub finansowych relacji, które mogłyby być interpretowane jako potencjalny konflikt interesów.
Brown, A. L., i Mccarthy, R. E. (1974). Relationship between measles and canine distemper viruses determined by delayed type hypersensitivity reactions in dogs. Nature 248, 344-345. doi: 10.1038/248344a0
PubMed Abstract | CrossRef Full Text | Google Scholar
.