Morbillivirus são patógenos altamente contagiosos e são responsáveis por vários surtos em populações não expostas (Pfeffermann et al., 2018). Eles pertencem à ordem Mononegavirales e família Paramyxoviridae e são caracterizados por um genoma RNA não segmentado, linear, de cadeia negativa (Lamb and Parks, 2013). Os morbillivírus distinguem-se por causar doenças respiratórias, gastrointestinais, imunossupressão e/ou neurológicas moderadas a graves numa vasta gama de hospedeiros, incluindo humanos (vírus do sarampo), carnívoros (morbillivírus canino anteriormente vírus da doença canina), bovinos (vírus da peste bovina), golfinhos e botos, e outras espécies selvagens em perigo (Lamb and Parks, 2013; Martinez-Gutierrez e Ruiz-Saenz, 2016).
O vírus das Maçãs (MeV) e o morbillivírus canino (CDV) são considerados os vírus mais contagiosos desta família (De Vries et al.., 2015), e devido ao alto potencial de transmissão do CDV, bem como ao seu potencial de transmissão entre espécies, a saúde global e as autoridades conservacionistas estão muito preocupadas com o papel do CDV na conservação de espécies ameaçadas e o possível “salto” de animais para humanos (Terio and Craft, 2013; Ohishi et al., 2014). Os cães domésticos são o principal hospedeiro dos CDV e também poderiam ser considerados como um reservatório para outros mamíferos (Suzuki et al., 2015; Duque-valencia et al., 2019); entretanto, com base na biologia dos CDV, os humanos também poderiam se tornar um alvo potencial (Cosby e Weir, 2018; Rendon-Marin et al., 2019).
Tentando entender o risco potencial de transmissão do VDC para os humanos, é necessário reunir todas as evidências existentes; e o estudo da origem e disseminação deste agente na população canina poderia apresentar uma importante chave para a compreensão deste processo. Recentemente, um artigo publicado no International Journal of Paleopathology convidou para uma discussão sobre a origem evolutiva do VDC. Ele conclui que o VDC se originou como um patógeno pandêmico na América do Sul após a infecção e adaptação do MeV a cães durante o período de colonização sul-americana. Este resultado foi obtido através de uma abordagem interdisciplinar adotada pela sintetização de uma análise paleopatológica de 96 cães pré-colombianos (750-1470 d.C.) da cidade velha de Weyanoke, Virginia site, com relatórios históricos, análise molecular e epidemiologia morbilliviral (Uhl et al, 2019).
Notadamente, populações caninas nativas da América quase desapareceram após o período de colonização, e cães europeus e eurasianos foram introduzidos no continente, deixando pouca herança genética de seus predecessores americanos (Ni Leathlobhair et al., 2018). Outro fator importante a ser considerado é que doenças “desconhecidas” também poderiam ter sido introduzidas, tornando mais difícil o rastreamento da origem de novos patógenos. Além disso, a pressão de seleção artificial sobre os cães domésticos e até mesmo sobre as populações humanas, particularmente durante o período de colonização, poderia ter aumentado a incidência de doenças, limitando assim a variação genética (Ostrander et al., 2017), o que por sua vez poderia significar uma resposta menos efetiva contra patógenos.
entre esses “novos” patógenos/doenças, o CDV foi descrito pela primeira vez por Antonio de Ulloa y de la Torre-Giral em 1746 como uma doença que afetava cães na região de Quito e nas outras partes da América do Sul, e foi relatado logo em seguida na Europa. O CDV foi registrado na Espanha em 1760, com 900 mortes ocorridas em um único dia em Madri, e 3 anos depois, ou seja, em 1764 e 1770, tinha chegado à Grã-Bretanha e Itália, respectivamente (Blancou, 2004). A transmissibilidade de vírus e maior suscetibilidade dos cachorros em comparação com cães adultos foram relatados mais tarde por Edward Jenner no início do século XIX. Ele comparou sua transmissibilidade com a do MeV e descobriu que os sobreviventes estavam protegidos de infecções subseqüentes (Jenner, 1809; Nambulli et al., 2016).
Briefly, após a chegada dos pioneiros europeus no século XV, novas doenças infecciosas se tornaram indiscutivelmente a consequência mais devastadora da colonização porque as populações indígenas americanas não tinham nenhuma exposição prévia a patógenos que tinham se tornado comuns na Europa (Walker et al., 2015). As epidemias múltiplas de sarampo, portanto, devastaram as populações indígenas americanas (Walker et al., 2015; Nambulli et al., 2016). Uhl et al., através de uma abordagem mista de evidências paleopatológicas, históricas, moleculares e epidemiológicas, relataram que epidemias graves de MeV nas populações indígenas americanas facilitaram o salto do MeV para grandes populações caninas domésticas de ambientes urbanos na América do Sul e a adaptação do vírus como CDV endêmico (Uhl et al., 2019). Além disso, registros históricos puderam provar que poucos anos após essa adaptação aos cães sul-americanos, o CDV foi transportado para a Europa em 1760, onde inicialmente induziu epidemias generalizadas com alta mortalidade antes de se tornar endêmico (Jenner, 1809).
No entanto, a filogeografia molecular relacionada às previsões evolutivas e à época do mais recente ancestral comum (tMRCA) foi calculada para a origem do VDC nos Estados Unidos na década de 1880 (95% de maior densidade posterior, 1858-1913) (Panzera et al, 2015), o que contradiz claramente a descrição do vírus na Europa no século XVIII. As análises de sequências que levaram a esta hipótese devem ser cuidadosamente examinadas devido ao viés e à disponibilidade limitada das sequências que foram utilizadas nesta reconstrução da filogeografia molecular. Além disso, muitas sequências ancestrais originais foram perdidas devido à capacidade do genoma viral do RNA do CDV e outros morbillivírus. Estes fatores deram origem ao questionamento da utilidade dos cálculos atuais do tMRCA para os vírus RNA (Sharp e Simmonds, 2011; Nambulli et al., 2016).
De acordo com Uhl et al, o morbillivírus poderia ter originado de bovinos por volta de 376 AC no “velho continente” (Figura 1), e a domesticação de animais pode ter tido uma influência significativa em eventos de espécies cruzadas, provavelmente traçando um ponto de partida no surgimento do MeV até aproximadamente 900 AC (Uhl et al., 2019). Ao contrário das atuais reconstruções filogenéticas CDV, a divergência do MeV é fortemente apoiada pela análise filogenética Bayesiana do relógio relaxado. O tempo de divergência entre o MeV e o vírus da peste bovina tinha sido demonstrado em aproximadamente os séculos XI a XII (Furuse et al., 2010). Outros dados moleculares, como a presença de um novo morbillivírus (intimamente relacionado ao CDV e PDV) circulando em morcegos do Brasil (DrMV), permitem especular que o CDV e o DrMV possam compartilhar um ancestral sul-americano comum (Drexler et al., 2012), apoiando assim indiretamente a idéia da origem sul-americana inicial do CDV.
>
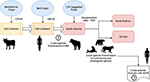
Figura 1. Representação esquemática da possível via de transmissão evolutiva do morbillivírus canino (CDV). Ver texto para referências.
Além do significado epistemológico e/ou científico da origem geográfica e data da divergência dos CDV, existem pistas importantes que devem ser esclarecidas para melhor entender o impacto atual dos CDV na transmissão interespécies, conservação animal e potencial zoonótico (Figura 1). É claro que, ao contrário da infecção pelo MeV, que é mantida por um único hospedeiro (humanos), o CDV tem sido amplamente comprovado como uma infecção/doença promíscua causadora de patógenos em uma vasta gama de espécies carnívoras e não carnívoras (Martinez-Gutierrez e Ruiz-Saenz, 2016). Esta promiscuidade tem sido atribuída não apenas à capacidade da hemaglutinina CDV (H) de interagir com receptores celulares hospedeiros, como o SLAM em células mononucleares e a nectina-4 em células epiteliais, mas também à semelhança entre as sequências de espécies dos receptores acima mencionados (Rendon-Marin et al., 2019). A similaridade de aminoácidos entre os receptores SLAM de mamíferos, incluindo mamíferos marinhos, é >80% (Ohishi et al., 2014), suportando assim os resultados da transmissão entre espécies. Além disso, há uma falta de variação relacionada à espécie nas sequências de nectina-4 entre humanos, ratos e cães, porque a nectina-4 humana poderia funcionar como um receptor in vitro para CDV (Noyce et al., 2011).
Epidemias naturais de CDV em diferentes primatas não humanos levantaram uma preocupação com a possível transmissão de CDV para humanos (Yoshikawa et al., 1989; Sun et al., 2010; Qiu et al., 2011; Sakai et al., 2013a). Há relatos de que as cepas de macacos CDV têm a capacidade intrínseca de usar a nectina-4 humana para a entrada do vírus e que esses CDV macacos se adaptam facilmente ao uso do receptor CD150 humano (SLAM) após mudanças mínimas de aminoácidos na proteína H viral (Bieringer et al., 2013; Sakai et al., 2013b). Entretanto, com base na infecção in vivo experimental por CDV de Cynomolgus macaques (Macaca fascicularis) na presença da imunidade MeV, os macacos foram parcialmente protegidos do desafio CDV (De Vries et al., 2014). Isto sugere que embora o VCD possa prontamente infectar primatas, a imunidade do MeV é protetora e que a infecção pelo VCD pode ser autolimitada. Transferindo este resultado para humanos, existe um risco potencial de infecção pelo VDC em pessoas que não possuem imunidade ao MeV de proteção cruzada devido à não vacinação e falhas na vacinação (Haralambieva et al., 2015) ou devido à ausência de vacinação na possível era pós-eradicação (Holzmann et al., 2016).
“Vírus emergentes” poderiam surgir através da transmissão cruzada de vírus de animais para humanos (Wolfe et al., 2007). Novos estudos, tanto estruturais quanto bioinformáticos, sugerem que apenas uma única mudança de aminoácidos em uma seqüência proteica poderia ser suficiente para superar a restrição no uso de receptores celulares entre dois hospedeiros diferentes, como humanos e ruminantes (Abdullah et al., 2018). Uma mutação única na proteína CDV H in vitro permite que este patógeno infecte células que expressam o receptor humano SLAM (Otsuki et al., 2013). Além disso, se abraçarmos a hipótese de que o CDV evoluiu do MeV, poderia ser possível que um descendente de CDV pudesse ser capaz de reinfectar humanos devido à evolução contínua tanto do vírus como de humanos, como foi previamente sugerido em outros modelos, mesmo que o ancestral “jumper virus” tivesse desaparecido da Terra há tempos (Emerman e Malik, 2010).
Outrossim, um dos resultados mais interessantes apresentados por Uhl et al. é a otimização dos genes CDV e MeV para o viés de uso do códão humano (CUB), sugerindo que o uso do códão CDV é mais próximo do CUB humano do que do CUB canino, porque o vírus ou seu progenitor, provavelmente MeV, foi inicialmente adaptado aos humanos (Uhl et al, 2019). CUB refere-se ao fenómeno em que alguns códões sinónimos são utilizados com mais frequência do que outros e como esta preferência varia dentro e entre espécies (Behura e Severson, 2013). Nos vírus RNA, o uso do códão está sob seleção porque os vírus são completamente dependentes dos tRNAs do hospedeiro e o viés resulta de vírus correspondentes ao uso do códão de seus hospedeiros (Jenkins e Holmes, 2003). A evolução pode às vezes favorecer os vírus que correspondem ao uso do códon do hospedeiro para promover a velocidade de replicação e adaptação ao hospedeiro como tem sido relatado em outros vírus RNA (Goni et al., 2012; Lauring et al., 2012; Di Paola et al., 2018; Freire et al., 2018).
Finalmente, gostaríamos de argumentar que alguns outros fatores devem ser considerados no possível cenário zoonótico do CDV. A neutralização cruzada entre MeV e CDV tem sido reconhecida desde muitos anos (Brown e Mccarthy, 1974), e esta premissa existe há mais de meio século quando a vacina do MeV foi usada para proteger os filhotes contra o CDV em uma idade em que a imunidade materna passiva freqüentemente interferia na vacinação contra o CDV (Baker et al., 1966; Brown et al., 1972). No entanto, o uso de uma vacina comercial dupla CDV/MeV ainda é recomendado para vacinação na presença de imunidade materna, e a vacina tem sido útil contra a doença clínica do sarampo em primatas não humanos (Christe et al., 2019). Assim, pode-se especular que a imunidade do rebanho MeV evita o salto de CDV e possível readaptação para humanos através da transmissão através de cães ou animais selvagens.
Comentários Finais
A evolução e origem dos patógenos virais não pode ser facilmente estudada; a partir daí, uma abordagem multidisciplinar é necessária para entender e talvez prever novas possíveis ameaças virais para humanos. Devido à sua biologia peculiar, patógenos virais como o CDV representam um modelo único para compreender os saltos entre espécies e o potencial zoonótico dos agentes virais muito próximo da população humana. Além dos tradicionais estudos filogenéticos moleculares e dos trabalhos de paleopatologia, os pesquisadores devem adotar diferentes abordagens para estudar a origem do CDV e os requisitos atuais de vírus e hospedeiro para os saltos interespécies. A introdução de métodos computacionais, como os estudos de bioinformática estrutural e paleovirologia, poderia ajudar na previsão e prevenção ou pelo menos proporcionar uma melhor compreensão desta doença emergente, e talvez, zoonótica de uma perspectiva diferente, considerando não apenas os dados seqüenciais, mas também as estruturas e funções como informação chave para este objetivo.
Contribuições dos autores
Todos os autores listados fizeram uma contribuição substancial, direta e intelectual ao trabalho, e o aprovaram para publicação.
Financiamento
Este trabalho foi apoiado financeiramente pelo Departamento Administrativo de Ciencia, Tecnología e Innovación-COLCIENCIAS Grant No. 123171249669 a JR-S.
Conflict of Interest Statement
Os autores declaram que a pesquisa foi realizada na ausência de quaisquer relações comerciais ou financeiras que pudessem ser interpretadas como um potencial conflito de interesses.
Brown, A. L., e Mccarthy, R. E. (1974). Relação entre sarampo e vírus da distemperose canina determinada por reações de hipersensibilidade de tipo retardado em cães. Nature 248, 344-345. doi: 10.1038/248344a0
PubMed Abstract | CrossRef Full Text | Google Scholar