La lipophilie est probablement la propriété physico-chimique la plus importante d’un médicament potentiel, elle joue un rôle dans la solubilité, l’absorption, la pénétration membranaire, la liaison aux protéines plasmatiques, la distribution, la pénétration dans le SNC et la partition dans d’autres tissus ou organes tels que le foie et a un impact sur les voies de clairance. Il est important dans la reconnaissance du ligand, non seulement à la protéine cible mais aussi aux interactions CYP450, à la liaison HERG et à l’induction enzymatique médiée par le PXR.
LogP est un composant de la règle des 5 de Lipinski une règle empirique pour prédire la solubilité et la perméabilité qui est devenue un substitut pour la similitude des médicaments.
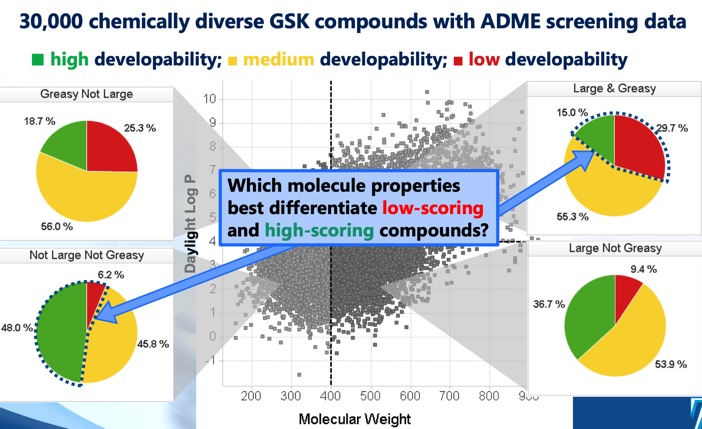
Le score de développabilité DOI identifie quatre régions distinctes cLog P/poids moléculaire qui définissent l’espace chimique optimal et sous-optimal, ainsi qu’un score de développabilité dérivé de modèles de régression utilisant les données de criblage de solubilité, de perméabilité, de liaison aux protéines et d’inhibition 3A4. Alors que le secteur MWt <400, cLogP <4 suggérait la plus grande chance de succès, il a été noté que même le secteur MWt >400, cLogP >4 comprenait quelques molécules développables bien qu’avec une chance de succès beaucoup plus faible.
La mesure de la lipophilie la plus couramment utilisée est le LogP, il s’agit du coefficient de partage d’une molécule entre une phase aqueuse et une phase lipophile, généralement l’octanol et l’eau.
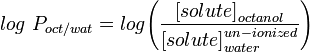
La mesure du LogP peut être entreprise de différentes manières, la plus courante est la méthode du shake-flask, qui consiste à dissoudre une partie du soluté en question dans un volume d’octanol et d’eau, à agiter pendant un certain temps, puis à mesurer la concentration du soluté dans chaque solvant. Cette méthode peut prendre beaucoup de temps, surtout s’il n’existe pas de méthode spectroscopique rapide pour mesurer la concentration de la molécule dans les phases. Une méthode plus rapide de détermination du log P fait appel à la chromatographie liquide à haute performance. Le log P d’un soluté peut être déterminé en corrélant son temps de rétention avec des composés similaires dont la valeur log P est connue doi.
Calcul de la lipophilie
En général, il n’est pas pratique de déterminer expérimentalement le LogP de chaque composé fabriqué (et il peut être intéressant de calculer le logP avant la synthèse) et donc les résultats calculés sont utilisés, il existe un certain nombre d’outils logiciels disponibles à la fois de bureau et en ligne (ne pas utiliser pour les composés confidentiels).
Plusieurs de ces applications fonctionnent en utilisant un grand ensemble de données d’entraînement de valeurs connues pour déterminer les contributions des fragments pour les sous-structures et les groupes fonctionnels, cependant le logP n’est pas une propriété additive simple et des termes de correction sont nécessaires pour permettre les effets de proximité, les liaisons H, les effets électroniques, etc. comme le montrent les exemples ci-dessous.
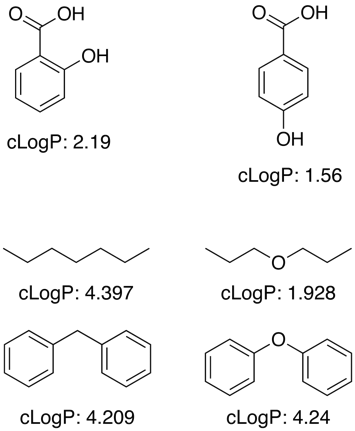
Pour les groupes fonctionnels inconnus, les programmes font souvent une approximation en utilisant les contributions des atomes individuels.
Les différentes méthodologies pour calculer le logP peuvent être divisées en trois approches différentes.
Atomique (par ex. « AlogP », ) &Atomique améliorée / Hybride (« XlogP », « SlogP »)
Fragment (« ClogP », KlogP, ACD/logP)
Méthodes basées sur les propriétés (« MlogP », « VlogP », « MClogP », « TlogP »)
Le logP atomique considère que chaque atome a une contribution au logP, et que les contributions à la valeur finale sont purement additives. Cependant, il est clair qu’un azote dans un amide est différent d’un azote dans une amine ou une pyridine, Amélioré Atomique prend en compte le type d’atome.
Les méthodes par fragments utilisent un grand ensemble de données d’entraînement de valeurs connues pour déterminer les contributions des fragments pour les sous-structures et les groupes fonctionnels, ainsi que des termes de correction pour tenir compte des effets de proximité. Ces méthodes se rabattent souvent sur des modèles atomiques pour les nouveaux groupes fonctionnels.
Les méthodes basées sur les propriétés ont tendance à être exigeantes en termes de calcul et ne sont pas vraiment adaptées pour tester de grands ensembles de données.
Parce que les ensembles d’entraînement et les algorithmes varient entre les applications, il est très important de ne pas combiner les résultats calculés en utilisant différents outils.
Certains outils permettent à l’utilisateur d’étendre l’ensemble d’entraînement en utilisant des valeurs mesurées en interne, cela peut être critique lors de l’exploration de nouveaux groupes fonctionnels ou d’échafaudages.
LogD
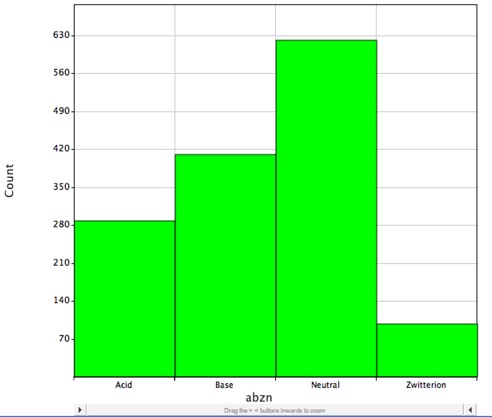
Cependant, la majorité des médicaments connus contiennent des groupes ionisables, comme le montre l’histogramme ci-dessous, cela montre la distribution des médicaments à petites molécules avec DrugBank et sont susceptibles d’être chargés au pH physiologique et LogP ne décrit correctement que le coefficient de partage des molécules neutres (non chargées).
LogD la constante de distribution est un meilleur descripteur de la lipophilie d’une molécule. Elle peut être déterminée de manière similaire à LogP mais au lieu d’utiliser de l’eau, la phase aqueuse est ajustée à un pH spécifique en utilisant un tampon. Le log D dépend donc du pH, d’où la nécessité de préciser le pH auquel le log D a été mesuré. Le log D à pH = 7,4 (le pH physiologique du sérum sanguin) est particulièrement intéressant.
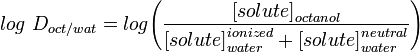
Des applications comme Marvin permettent à l’utilisateur de calculer le log D mais aussi d’afficher le profil de distribution du pH, comme indiqué ci-dessous pour la Warfarine.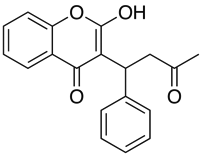
Pour les composés dont le pKa est proche du pH physiologique, il peut être critique de considérer ce qui pourrait réellement être la forme ionisée prédominante.
Cela peut également être précieux lorsqu’on pense à l’absorption à partir des différentes régions du canal alimentaire où le pH varie de 1-3 dans l’estomac à 7-8 dans l’iléon.
Les contributions de divers groupes fonctionnels au LogD ont été explorées « LogD contributions of substituents commonly used in medicinal chemistry » DOI, cette étude a utilisé l’analyse des paires moléculaires appariées des valeurs expérimentales de LogD de plusieurs milliers de composés collectés par la méthode du shake-flask à pH = 7,4. Ils ont rapporté la différence deltaLogD moyenne pour des paires moléculaires particulières et les résultats sont présentés ci-dessous pour le cas où le groupe fonctionnel se trouve à n’importe quelle position sur le cycle phényle. J’ai également inclus le LogD calculé en utilisant le logiciel Chemaxon.
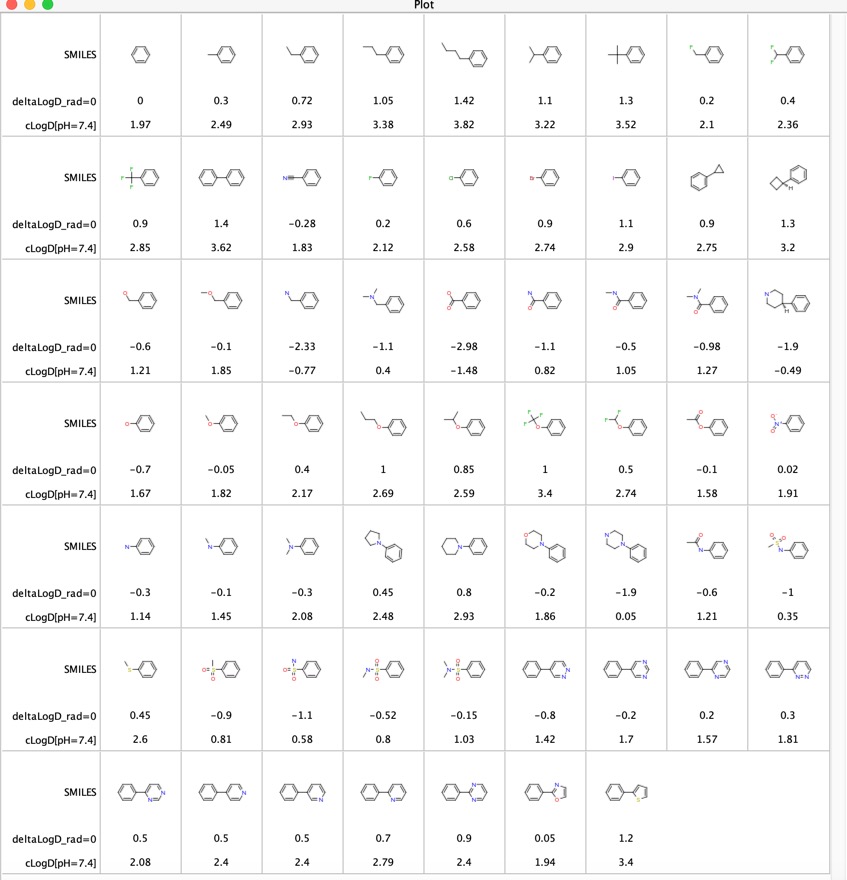
Ce tableau est utile pour comparer les groupes fonctionnels, en particulier les 11 dernières entrées comparent l’influence de divers hétérocycles ont sur le LogD. Ces hétérocycles sont souvent utilisés comme remplacements bioisostériques pour un cycle phényle.
J’ai pensé qu’il pourrait être intéressant de comparer les différences de LogD déterminées en utilisant les paires moléculaires appariées (deltaLogD_rad=0) avec les valeurs déterminées en utilisant le LogD calculé par Chemaxon (deltacLogD). Comme vous pouvez le voir ci-dessous, il y a une assez bonne correspondance.
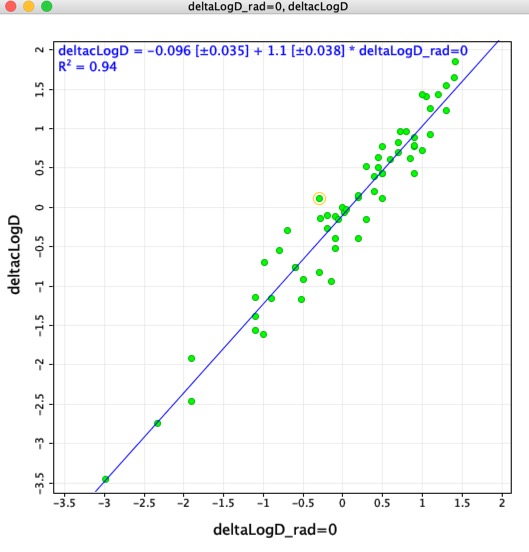
Rappellez-vous Parce que les ensembles d’entraînement et les algorithmes varient entre les applications, il est très important de ne pas combiner les résultats calculés à l’aide de différents outils.
Il est important d’être circonspect sur le fait que toute amélioration de l’affinité de liaison n’est pas entièrement dirigée par une augmentation du LogD, il est souvent utile de simplement tracer l’affinité de liaison en fonction du LogD. Les modifications de composés les plus intéressantes ne sont pas nécessairement celles qui donnent la plus grande augmentation de l’affinité, mais peuvent être celles qui donnent une augmentation de l’affinité sans une augmentation correspondante de la lipophilie. En regardant le tableau ci-dessous, il y a un certain nombre de composés à très haute affinité.
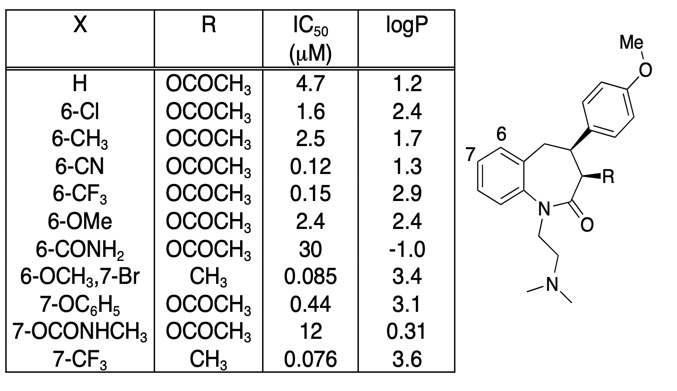
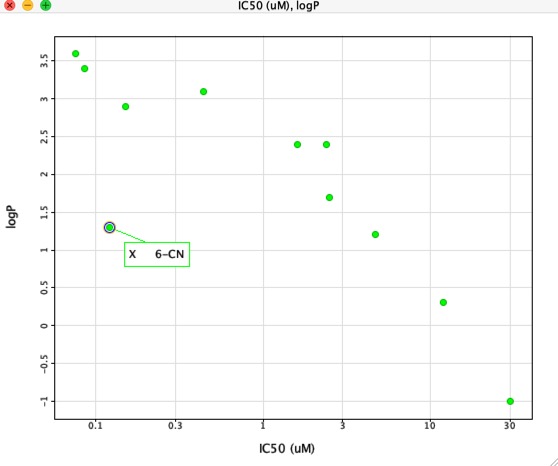
Cependant, si nous traçons la CI50 en fonction du LogP comme indiqué ci-dessous, il y a une corrélation très claire entre le LogP et la CI50, cependant un composé est clairement différent. Le substituant 6-CN donne une augmentation de l’affinité sans une augmentation correspondante du LogP.
La lipophilie est également un composant important beaucoup des responsabilités hors cible, y compris la liaison aux protéines plasmatiques (en particulier l’albumine), HERG, les interactions CYP, les transporteurs, ont de fortes corrélations avec la lipophilie, et il y a eu un certain nombre d’études reliant un logP élevé à la probabilité que les composés échouent dans le développement en raison de mauvaises caractéristiques ADMET (absorption, distribution, métabolisme, excrétion et toxicité). En revanche, il est souvent évident qu’une certaine taille et une certaine lipophilie sont nécessaires pour obtenir des niveaux raisonnables d’affinité. Trouver un équilibre entre ces exigences est un défi majeur dans la découverte de médicaments et il est suggéré aux chimistes de cibler le point idéal, à savoir un MWt de 250 à 500 et un LogP de 2 à 4. Une conséquence de cette approche est la nécessité de donner la priorité aux composés de faible poids moléculaire et moins lipophiles lors du criblage. L’objectif initial de la chimie médicinale devrait être de sélectionner des points de départ de bonne qualité, puis de contrôler efficacement les changements de propriétés physicochimiques au cours du processus d’optimisation.
Mérite d’être lu
Finding the sweet spot : the role of nature and nurture in medicinal chemistry DOI
Lipophilic efficiency : the most important efficiency metric in medicinal chemistry DOI
Dernière mise à jour le 12 janvier 2019
.