Les morbillivirus sont des agents pathogènes hautement contagieux et sont responsables de diverses épidémies dans des populations non exposées (Pfeffermann et al., 2018). Ils appartiennent à l’ordre des Mononegavirales et à la famille des Paramyxoviridae et sont caractérisés par un génome ARN non segmenté, linéaire et à brin négatif (Lamb et Parks, 2013). Les morbillivirus se distinguent pour causer des maladies respiratoires, gastro-intestinales, immunosuppressives et/ou neurologiques modérées à sévères chez un large éventail d’hôtes, notamment les humains (virus de la rougeole), les carnivores (morbillivirus canin anciennement virus de la maladie de Carré), le bétail (virus de la peste bovine), les dauphins et les marsouins, ainsi que d’autres espèces sauvages menacées (Lamb et Parks, 2013 ; Martinez-Gutierrez et Ruiz-Saenz, 2016).
Le virus de la rougeole (MeV) et le morbillivirus canin (CDV) sont considérés comme les virus les plus contagieux parmi cette famille (De Vries et al…, 2015), et en raison du fort potentiel de transmission du CDV ainsi que de son potentiel de transmission inter-espèces, les autorités sanitaires mondiales et les défenseurs de l’environnement sont très préoccupés par le rôle du CDV sur la conservation des espèces menacées et le possible » saut » des animaux aux humains (Terio et Craft, 2013 ; Ohishi et al., 2014). Les chiens domestiques sont le principal hôte du CDV et pourraient également être considérés comme un réservoir pour d’autres mammifères (Suzuki et al., 2015 ; Duque-valencia et al., 2019) ; cependant, sur la base de la biologie du CDV, les humains pourraient également se transformer en une cible potentielle (Cosby et Weir, 2018 ; Rendon-Marin et al, 2019).
Pour tenter de comprendre le risque potentiel de transmission du CDV à l’homme, il est nécessaire de rassembler toutes les preuves existantes ; et l’étude de l’origine et de la dissémination de cet agent dans la population canine pourrait présenter une clé importante pour comprendre ce processus. Récemment, un article publié dans l’International Journal of Paleopathology a invité à une discussion sur l’origine évolutive du CDV. Il conclut que le CDV est né en tant qu’agent pathogène pandémique en Amérique du Sud suite à l’infection et à l’adaptation du MeV aux chiens pendant la période de colonisation de l’Amérique du Sud. Ce résultat a été obtenu via une approche interdisciplinaire adoptée en synthétisant une analyse paléopathologique de 96 chiens précolombiens (750-1470 CE) provenant du site de Weyanoke Old Town, en Virginie, avec des rapports historiques, une analyse moléculaire et une épidémiologie morbillivirale (Uhl et al, 2019).
Notamment, les populations de chiens indigènes d’Amérique ont presque disparu après la période de colonisation, et les chiens européens et eurasiens ont été introduits sur le continent, laissant peu de fond génétique de ses prédécesseurs américains (Ni Leathlobhair et al., 2018). Un autre facteur important à prendre en compte est que des maladies » inconnues » ont également pu être introduites, ce qui rend plus difficile le suivi de l’origine des nouveaux agents pathogènes. En outre, la pression de sélection artificielle sur les chiens domestiques et même sur les populations humaines, en particulier pendant la période de colonisation, pourrait avoir renforcé l’incidence des maladies, limitant ainsi la variation génétique (Ostrander et al, 2017), ce qui à son tour pourrait signifier une réponse moins efficace contre les agents pathogènes.
Parmi ces « nouveaux » agents pathogènes/maladies, le CDV a été décrit pour la première fois par Antonio de Ulloa y de la Torre-Giral en 1746 comme une maladie affectant les chiens dans la région de Quito et dans les autres parties de l’Amérique du Sud, et il a été signalé peu après en Europe. Le CDV a été enregistré en Espagne en 1760, avec 900 décès en un seul jour à Madrid, et 3 ans plus tard, c’est-à-dire en 1764 et 1770, il avait atteint la Grande-Bretagne et l’Italie, respectivement (Blancou, 2004). La transmissibilité du virus et la plus grande susceptibilité des chiots par rapport aux chiens adultes ont été rapportées par Edward Jenner au début des années 1800. Il a comparé leur transmissibilité à celle des MeV et a découvert que les survivants étaient protégés contre une infection ultérieure (Jenner, 1809 ; Nambulli et al., 2016).
Brièvement, après l’arrivée des pionniers européens au XVe siècle, les nouvelles maladies infectieuses sont sans doute devenues la conséquence la plus dévastatrice de la colonisation, car les populations américaines indigènes n’avaient aucune exposition préalable aux agents pathogènes devenus courants en Europe (Walker et al., 2015). De multiples épidémies de rougeole ont donc dévasté les populations autochtones américaines (Walker et al., 2015 ; Nambulli et al., 2016). Uhl et al. via une approche mixte de preuves paléopathologiques, historiques, moléculaires et épidémiologiques, ont rapporté que de graves épidémies de VME dans les populations indigènes américaines ont facilité le saut du VME vers les grandes populations de chiens domestiques des environnements urbains en Amérique du Sud et l’adaptation du virus en tant que VCD endémique (Uhl et al., 2019). De plus, les archives historiques pourraient prouver que quelques années après cette adaptation aux chiens sud-américains, le CDV a été transporté en Europe en 1760, où il a d’abord induit des épidémies généralisées avec une mortalité élevée avant de devenir endémique (Jenner, 1809).
Cependant, la phylogéographie moléculaire liée aux prédictions évolutives et le temps jusqu’à l’ancêtre commun le plus récent (tMRCA) ont été calculés pour l’origine du CDV aux États-Unis dans les années 1880 (densité postérieure la plus élevée à 95%, 1858-1913) (Panzera et al, 2015), ce qui contredit clairement la description du virus en Europe au XVIIIe siècle. Les analyses de séquences qui ont conduit à cette hypothèse doivent être soigneusement examinées en raison du biais et de la disponibilité limitée des séquences qui ont été utilisées dans cette reconstruction de phylogéographie moléculaire. De plus, de nombreuses séquences ancestrales originales ont été perdues en raison de la labilité du génome de l’ARN viral du CDV et d’autres morbillivirus. Ces facteurs ont donné lieu à la remise en question de l’utilité des calculs actuels du tMRCA pour les virus à ARN (Sharp et Simmonds, 2011 ; Nambulli et al., 2016).
Selon Uhl et al, morbillivirus pourrait être originaire des bovins vers 376 AC dans le « vieux continent » (Figure 1), et la domestication des animaux peut avoir eu une influence significative sur les événements inter-espèces, traçant probablement un point de départ dans l’émergence de MeV à environ 900 AC (Uhl et al., 2019). Contrairement aux reconstructions phylogénétiques actuelles du CDV, la divergence de MeV est fortement soutenue par l’analyse phylogénétique bayésienne à horloge relaxée. Il a été démontré que le moment de la divergence entre le MeV et le virus de la peste bovine s’est produit approximativement entre le XIe et le XIIe siècle (Furuse et al., 2010). D’autres données moléculaires, telles que la présence d’un nouveau morbillivirus (étroitement apparenté au CDV et au PDV) circulant chez les chauves-souris du Brésil (DrMV), permettent de spéculer que le CDV et le DrMV pourraient partager un ancêtre sud-américain commun (Drexler et al., 2012), soutenant ainsi indirectement l’idée d’une origine sud-américaine précoce du CDV.
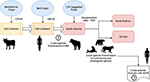
Figure 1. Représentation schématique de la voie de transmission évolutive possible du morbillivirus canin (CDV). Voir le texte pour les références.
Au delà de la signification épistémologique et/ou scientifique de l’origine géographique et de la date de divergence du CDV, il existe des indices importants qui doivent être clarifiés pour mieux comprendre l’impact actuel des CDV sur la transmission inter-espèces, la conservation des animaux et le potentiel zoonotique (Figure 1). Il est clair que, contrairement à l’infection par le MeV, qui est maintenue par un seul hôte (l’homme), il a été largement prouvé que le CDV est un pathogène promiscuous causant des infections/maladies chez une vaste gamme d’espèces carnivores et non carnivores (Martinez-Gutierrez et Ruiz-Saenz, 2016). Cette promiscuité a été attribuée non seulement à la capacité de l’hémagglutinine (H) du CDV à interagir avec les récepteurs cellulaires de l’hôte, tels que le SLAM dans les cellules mononucléaires et la nectine-4 dans les cellules épithéliales, mais aussi à la similarité entre les séquences des espèces des récepteurs mentionnés ci-dessus (Rendon-Marin et al., 2019). La similitude des acides aminés entre les récepteurs SLAM des mammifères, y compris les mammifères marins, est >80 % (Ohishi et al., 2014), ce qui étaye les résultats de la transmission inter-espèces. En outre, il y a un manque de variation liée à l’espèce dans les séquences de nectine-4 chez les humains, les souris et les chiens parce que la nectine-4 humaine pourrait fonctionner comme un récepteur in vitro pour le CDV (Noyce et al., 2011).
Les épidémies naturelles de CDV chez différents primates non humains ont soulevé une préoccupation concernant la transmission possible du CDV aux humains (Yoshikawa et al., 1989 ; Sun et al., 2010 ; Qiu et al., 2011 ; Sakai et al., 2013a). Certains rapports indiquent que les souches de singe du CDV ont la capacité intrinsèque d’utiliser la nectine-4 humaine pour l’entrée du virus et que ces singes CDV s’adaptent facilement pour utiliser le récepteur CD150 (SLAM) humain après des modifications minimales des acides aminés de la protéine H virale (Bieringer et al., 2013 ; Sakai et al., 2013b). Cependant, d’après l’infection expérimentale in vivo par le CDV de macaques de Cynomolgus (Macaca fascicularis) en présence d’une immunité MeV, les macaques ont bénéficié d’une protection croisée partielle contre le défi du CDV (De Vries et al., 2014). Cela suggère que, bien que le CDV puisse facilement infecter les primates, l’immunité MeV est protectrice et que l’infection par le CDV pourrait être autolimitée. En transférant ce résultat à l’homme, il existe un risque potentiel d’infection par le CDV chez les personnes qui n’ont pas d’immunité MeV de protection croisée en raison de la non-vaccination et des échecs vaccinaux (Haralambieva et al., 2015) ou en raison de l’absence de vaccination dans l’éventuelle ère post-éradication (Holzmann et al., 2016).
Les « virus émergents » pourraient apparaître via la transmission inter-espèces de virus des animaux vers l’homme (Wolfe et al., 2007). De nouvelles études, tant structurelles que bioinformatiques, suggèrent qu’un seul changement d’acide aminé dans une séquence protéique pourrait suffire à surmonter la restriction d’utilisation des récepteurs cellulaires entre deux hôtes différents, tels que les humains et les ruminants (Abdullah et al., 2018). Une mutation unique dans la protéine H du CDV in vitro permet à cet agent pathogène d’infecter des cellules exprimant le récepteur SLAM humain (Otsuki et al., 2013). En outre, si nous embrassons l’hypothèse selon laquelle le CDV a évolué à partir du MeV, il serait possible qu’un descendant du CDV soit capable de réinfecter les humains en raison de l’évolution continue à la fois du virus et des humains, comme cela a été suggéré précédemment dans d’autres modèles, même si le « virus sauteur » ancestral avait disparu de la terre il y a longtemps (Emerman et Malik, 2010).
De plus, l’un des résultats les plus intéressants présentés par Uhl et al. est l’optimisation des gènes CDV et MeV au biais d’utilisation des codons humains (CUB), ce qui suggère que l’utilisation des codons CDV est plus proche du CUB humain que du CUB canin parce que le virus ou son progéniteur, très probablement MeV, a été initialement adapté aux humains (Uhl et al…, 2019). Le CUB désigne le phénomène selon lequel certains codons synonymes sont utilisés plus souvent que d’autres et la façon dont cette préférence varie au sein d’une même espèce et entre espèces (Behura et Severson, 2013). Chez les virus à ARN, l’utilisation des codons fait l’objet d’une sélection parce que les virus sont complètement dépendants des ARNt de l’hôte et que le biais résulte du fait que les virus correspondent à l’utilisation des codons de leurs hôtes (Jenkins et Holmes, 2003). L’évolution peut parfois favoriser les virus qui correspondent à l’utilisation des codons de leur hôte pour promouvoir la vitesse de réplication et l’adaptation à l’hôte, comme cela a été signalé dans d’autres virus à ARN (Goni et al., 2012 ; Lauring et al., 2012 ; Di Paola et al., 2018 ; Freire et al., 2018).
Enfin, nous aimerions faire valoir que certains autres facteurs doivent être pris en compte dans le scénario zoonotique possible du CDV. La neutralisation croisée entre le MeV et le CDV est reconnue depuis de nombreuses années (Brown et Mccarthy, 1974), et cette prémisse existe depuis plus d’un demi-siècle lorsque le vaccin MeV a été utilisé pour protéger les chiots contre le CDV à un âge où l’immunité maternelle passive interférait souvent avec la vaccination contre le CDV (Baker et al., 1966 ; Brown et al., 1972). Néanmoins, l’utilisation d’un vaccin commercial double CDV/MeV est toujours recommandée pour la vaccination en présence d’une immunité maternelle, et le vaccin s’est avéré utile contre la maladie clinique de la rougeole chez les primates non humains (Christe et al., 2019). Par conséquent, on peut spéculer que l’immunité de troupeau MeV évite le saut du CDV et une éventuelle réadaptation aux humains via la transmission par les chiens ou les animaux sauvages.
Marques finales
L’évolution et l’origine des pathogènes viraux ne peuvent pas être facilement étudiées ; par la suite, une approche multidisciplinaire est nécessaire pour comprendre et peut-être prédire les nouvelles menaces virales possibles pour les humains. En raison de leur biologie particulière, les pathogènes viraux tels que le CDV représentent un modèle unique pour comprendre le saut inter-espèces et le potentiel zoonotique d’agents viraux très proches de la population humaine. Outre les études phylogénétiques moléculaires traditionnelles et les travaux de paléopathologie, les chercheurs doivent adopter différentes approches pour étudier l’origine du CDV et les exigences actuelles du virus et de l’hôte pour le saut inter-espèces. L’introduction de méthodes computationnelles, telles que la bioinformatique structurelle et les études de paléovirologie, pourrait aider à la prédiction et à la prévention ou du moins fournir une meilleure compréhension de cette maladie émergente, et peut-être, zoonotique d’un point de vue différent en considérant non seulement les données de séquençage mais aussi les structures et les fonctions comme des informations clés à cet objectif.
Contributions des auteurs
Tous les auteurs listés ont apporté une contribution substantielle, directe et intellectuelle au travail, et l’ont approuvé pour la publication.
Financement
Ce travail a été financièrement soutenu par le Departamento Administrativo de Ciencia, Tecnología e Innovación-COLCIENCIAS Subvention n° 123171249669 à JR-S.
Déclaration de conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière qui pourrait être interprétée comme un conflit d’intérêts potentiel.
Brown, A. L.., et Mccarthy, R. E. (1974). Relation entre les virus de la rougeole et de la maladie de Carré déterminée par des réactions d’hypersensibilité de type retardé chez le chien. Nature 248, 344-345. doi : 10.1038/248344a0
PubMed Abstract | CrossRef Full Text | Google Scholar