Los morbillivirus son patógenos altamente contagiosos y son responsables de diversos brotes en poblaciones no expuestas (Pfeffermann et al., 2018). Pertenecen al orden Mononegavirales y a la familia Paramyxoviridae y se caracterizan por tener un genoma de ARN no segmentado, lineal y de cadena negativa (Lamb y Parks, 2013). Los morbilivirus se distinguen por causar enfermedades respiratorias, gastrointestinales, de inmunosupresión y/o neurológicas de moderadas a graves en una amplia gama de huéspedes, incluyendo a los seres humanos (virus del sarampión), carnívoros (morbilivirus canino antes virus del moquillo canino), ganado (virus de la peste bovina), delfines y marsopas, y otras especies de fauna silvestre en peligro de extinción (Lamb y Parks, 2013; Martínez-Gutiérrez y Ruiz-Sáenz, 2016).
El virus del sarampión (MeV) y el morbillivirus canino (CDV) se consideran los virus más contagiosos entre esta familia (De Vries et al., 2015), y debido al alto potencial de transmisión del CDV, así como a su potencial de transmisión entre especies, las autoridades sanitarias y conservacionistas mundiales están muy preocupadas por el papel del CDV en la conservación de las especies en peligro de extinción y el posible «salto» de los animales a los seres humanos (Terio y Craft, 2013; Ohishi et al., 2014). Los perros domésticos son el principal huésped del CDV y también podrían ser considerados como un reservorio para otros mamíferos (Suzuki et al., 2015; Duque-valencia et al., 2019); sin embargo, basándose en la biología del CDV, los humanos también podrían convertirse en un objetivo potencial (Cosby y Weir, 2018; Rendon-Marin et al, 2019).
Para tratar de entender el riesgo potencial de transmisión del CDV a los humanos, es necesario reunir todas las evidencias existentes; y el estudio del origen y diseminación de este agente en la población canina podría presentar una clave importante para entender este proceso. Recientemente, un artículo publicado en el International Journal of Paleopathology invitaba a un debate sobre el origen evolutivo del CDV. En él se concluye que el CDV se originó como patógeno pandémico en Sudamérica tras la infección y adaptación del MeV a los perros durante el periodo de colonización sudamericana. Este resultado se obtuvo a través de un enfoque interdisciplinario adoptado al sintetizar un análisis paleopatológico de 96 perros precolombinos (750-1470 CE) del sitio de Weyanoke Old Town, Virginia, con informes históricos, análisis molecular y epidemiología morbilliviral (Uhl et al, 2019).
Cabe destacar que las poblaciones caninas autóctonas de América prácticamente desaparecieron tras el periodo de colonización, y que los perros europeos y euroasiáticos fueron introducidos en el continente, dejando pocos antecedentes genéticos de sus antecesores americanos (Ni Leathlobhair et al., 2018). Otro factor importante que vale la pena considerar es que también se podrían haber introducido enfermedades «desconocidas», lo que hace más difícil rastrear el origen de los nuevos patógenos. Además, la presión de selección artificial sobre los perros domésticos e incluso las poblaciones humanas, especialmente durante el periodo de colonización, podría haber aumentado la incidencia de las enfermedades, limitando así la variación genética (Ostrander et al., 2017), lo que a su vez podría significar una respuesta menos eficaz contra los patógenos.
Entre estos «nuevos» patógenos/enfermedades, el CDV fue descrito por primera vez por Antonio de Ulloa y de la Torre-Giral en 1746 como una enfermedad que afectaba a los perros en la región de Quito y en otras partes de Sudamérica, y se informó poco después en Europa. El CDV se registró en España en 1760, con 900 muertes en un solo día en Madrid, y 3 años después, es decir, en 1764 y 1770, había llegado a Gran Bretaña e Italia, respectivamente (Blancou, 2004). La transmisibilidad del virus y la mayor susceptibilidad de los cachorros en comparación con los perros adultos fueron señaladas posteriormente por Edward Jenner a principios de 1800. Comparó su transmisibilidad con la de los MeV y descubrió que los supervivientes estaban protegidos de la infección posterior (Jenner, 1809; Nambulli et al., 2016).
Brevemente, tras la llegada de los pioneros europeos en el siglo XV, las nuevas enfermedades infecciosas se convirtieron, posiblemente, en la consecuencia más devastadora de la colonización, ya que las poblaciones indígenas americanas no habían estado expuestas previamente a los patógenos que se habían hecho comunes en Europa (Walker et al., 2015). Por lo tanto, múltiples epidemias de sarampión devastaron a las poblaciones indígenas americanas (Walker et al., 2015; Nambulli et al., 2016). Uhl et al. mediante un enfoque mixto de pruebas paleopatológicas, históricas, moleculares y epidemiológicas, informaron de que las graves epidemias de VME en las poblaciones indígenas americanas facilitaron el salto del VME a las grandes poblaciones de perros domésticos de los entornos urbanos de Sudamérica y la adaptación del virus como VCD endémico (Uhl et al., 2019). Asimismo, los registros históricos podrían demostrar que pocos años después de esa adaptación a los perros sudamericanos, el CDV fue transportado a Europa en 1760, donde inicialmente indujo epidemias generalizadas con alta mortalidad antes de convertirse en endémico (Jenner, 1809).
Sin embargo, la filogeografía molecular relacionada con las predicciones evolutivas y el tiempo hasta el ancestro común más reciente (tMRCA) se calcularon para el origen del CDV en los Estados Unidos en la década de 1880 (95% de densidad posterior más alta, 1858-1913) (Panzera et al, 2015), lo que contradice claramente la descripción del virus en Europa en el siglo XVIII. Los análisis de secuencias que condujeron a esta hipótesis deben ser examinados cuidadosamente debido al sesgo y a la limitada disponibilidad de las secuencias que se utilizaron en esta reconstrucción filogeográfica molecular. Además, muchas secuencias ancestrales originales se han perdido debido a la labilidad del genoma del ARN viral del CDV y de otros morbillivirus. Estos factores han dado lugar al cuestionamiento de la utilidad de los cálculos actuales de tMRCA para los virus de ARN (Sharp y Simmonds, 2011; Nambulli et al., 2016).
Según Uhl et al, el morbillivirus podría haberse originado en el ganado alrededor del año 376 a.C. en el «viejo continente» (Figura 1), y la domesticación de los animales puede haber tenido una influencia significativa en los eventos de cruce de especies, lo que probablemente traza un punto de partida en la aparición del VME hasta aproximadamente el año 900 a.C. (Uhl et al., 2019). A diferencia de las reconstrucciones filogenéticas actuales de CDV, la divergencia de MeV está fuertemente apoyada por el análisis filogenético bayesiano de reloj relajado. Se ha demostrado que el tiempo de divergencia entre el VME y el virus de la peste bovina se produjo aproximadamente entre los siglos XI y XII (Furuse et al., 2010). Otros datos moleculares, como la presencia de un nuevo morbillivirus (estrechamente relacionado con el CDV y el PDV) que circula en murciélagos de Brasil (DrMV), permiten especular que el CDV y el DrMV podrían compartir un ancestro común sudamericano (Drexler et al., 2012), apoyando así indirectamente la idea del origen sudamericano temprano del CDV.
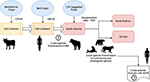
Figura 1. Representación esquemática de la posible ruta de transmisión evolutiva del morbillivirus canino (CDV). Véanse las referencias en el texto.
Más allá del significado epistemológico y/o científico del origen geográfico y la fecha de divergencia del CDV, hay pistas importantes que deben aclararse para comprender mejor el impacto actual de los CDV en la transmisión entre especies, la conservación de los animales y el potencial zoonótico (Figura 1). Está claro que, a diferencia de la infección por MeV, que se mantiene en un único huésped (los humanos), se ha demostrado ampliamente que el CDV es un patógeno promiscuo que causa infección/enfermedad en una amplia gama de especies carnívoras y no carnívoras (Martínez-Gutiérrez y Ruiz-Sáenz, 2016). Esta promiscuidad se ha atribuido no sólo a la capacidad de la hemaglutinina (H) del CDV para interactuar con los receptores celulares del huésped, como el SLAM en las células mononucleares y la nectina-4 en las células epiteliales, sino también a la similitud entre las secuencias de las especies de los receptores mencionados (Rendon-Marin et al., 2019). La similitud de aminoácidos entre los receptores SLAM de los mamíferos, incluidos los mamíferos marinos, es >80% (Ohishi et al., 2014), apoyando así los resultados de la transmisión entre especies. Además, hay una falta de variación relacionada con la especie en las secuencias de nectina-4 entre los seres humanos, los ratones y los perros porque la nectina-4 humana podría funcionar como un receptor in vitro para el CDV (Noyce et al., 2011).
Los brotes naturales de CDV en diferentes primates no humanos han planteado una preocupación con respecto a la posible transmisión del CDV a los seres humanos (Yoshikawa et al., 1989; Sun et al., 2010; Qiu et al., 2011; Sakai et al., 2013a). Hay informes que indican que las cepas de mono del CDV tienen la capacidad intrínseca de utilizar la nectina-4 humana para la entrada del virus y que esos CDV de mono se adaptan fácilmente para utilizar el receptor CD150 humano (SLAM) tras cambios mínimos de aminoácidos en la proteína H viral (Bieringer et al., 2013; Sakai et al., 2013b). Sin embargo, basándose en la infección experimental in vivo por el CDV de macacos Cynomolgus (Macaca fascicularis) en presencia de inmunidad MeV, los macacos estaban parcialmente protegidos de forma cruzada contra el desafío del CDV (De Vries et al., 2014). Esto sugiere que, aunque el CDV puede infectar fácilmente a los primates, la inmunidad MeV es protectora y que la infección por CDV podría ser autolimitada. Trasladando este resultado a los seres humanos, existe un riesgo potencial de infección por el CDV en personas que carecen de inmunidad cruzada contra el MeV debido a la no vacunación y a los fracasos de las vacunas (Haralambieva et al., 2015) o debido a la ausencia de vacunación en la posible era posterior a la erradicación (Holzmann et al., 2016).
Los «virus emergentes» podrían, según se informa, surgir a través de la transmisión entre especies de los virus de los animales a los seres humanos (Wolfe et al., 2007). Nuevos estudios, tanto estructurales como bioinformáticos, sugieren que un solo cambio de aminoácido en la secuencia de una proteína podría ser suficiente para superar la restricción en el uso de los receptores celulares entre dos huéspedes diferentes, como los humanos y los rumiantes (Abdullah et al., 2018). Una mutación única en la proteína H del CDV in vitro permite a este patógeno infectar células que expresan el receptor SLAM humano (Otsuki et al., 2013). Además, si abrazamos la hipótesis de que el CDV evolucionó a partir del MeV, podría ser posible que un descendiente del CDV fuera capaz de reinfectar a los humanos debido a la evolución continua tanto del virus como de los humanos, como se ha sugerido previamente en otros modelos aunque el «virus saltador» ancestral hubiera desaparecido de la Tierra hace tiempo (Emerman y Malik, 2010).
Además, uno de los resultados más interesantes presentados por Uhl et al. es la optimización tanto de los genes del CDV como del MeV al sesgo de uso de codones (CUB) humano, lo que sugiere que el uso de codones del CDV está más cerca del CUB humano que del CUB canino porque el virus o su progenitor, muy probablemente el MeV, se adaptó inicialmente a los humanos (Uhl et al., 2019). CUB se refiere al fenómeno en el que algunos codones sinónimos se utilizan con más frecuencia que otros y cómo esta preferencia varía dentro y entre las especies (Behura y Severson, 2013). En los virus de ARN, el uso de codones está bajo selección porque los virus son completamente dependientes de los ARNt del huésped y el sesgo resulta de que los virus coincidan con el uso de codones de sus huéspedes (Jenkins y Holmes, 2003). La evolución a veces puede favorecer a los virus que coinciden con el uso de codones de su huésped para promover la velocidad de replicación y la adaptación al huésped, como se ha informado en otros virus de ARN (Goni et al., 2012; Lauring et al., 2012; Di Paola et al., 2018; Freire et al., 2018).
Por último, nos gustaría argumentar que algunos otros factores deben ser considerados en el posible escenario zoonótico de CDV. La neutralización cruzada entre el MeV y el CDV ha sido reconocida desde hace muchos años (Brown y Mccarthy, 1974), y esta premisa ha existido durante más de medio siglo cuando la vacuna contra el MeV se utilizó para proteger a las crías contra el CDV en una edad en la que la inmunidad materna pasiva a menudo interfería con la vacunación contra el CDV (Baker et al., 1966; Brown et al., 1972). No obstante, se sigue recomendando el uso de una vacuna comercial dual CDV/MeV para la vacunación en presencia de inmunidad materna, y la vacuna ha sido útil contra la enfermedad clínica del sarampión en primates no humanos (Christe et al., 2019). Por lo tanto, se puede especular que la inmunidad de rebaño de MeV evita el salto de CDV y la posible readaptación a los seres humanos a través de la transmisión a través de perros o animales de la vida silvestre.
Observaciones finales
La evolución y el origen de los patógenos virales no se pueden estudiar fácilmente; en adelante, es necesario un enfoque multidisciplinario para comprender y tal vez predecir nuevas posibles amenazas virales para los seres humanos. Debido a su peculiar biología, los patógenos víricos como el CDV representan un modelo único para entender los saltos entre especies y el potencial zoonótico de agentes víricos muy cercanos a la población humana. Además de los tradicionales estudios filogenéticos moleculares y los trabajos de paleopatología, los investigadores deben adoptar diferentes enfoques para estudiar el origen del CDV y los actuales requisitos virales y del huésped para el salto interespecífico. La introducción de métodos computacionales, como la bioinformática estructural y los estudios de paleovirología, podría ayudar en la predicción y prevención o, al menos, proporcionar una mejor comprensión de esta enfermedad emergente, y tal vez zoonótica, desde una perspectiva diferente, considerando no sólo los datos de secuenciación, sino también las estructuras y funciones como información clave para este objetivo.
Contribuciones de los autores
Todos los autores enumerados han hecho una contribución sustancial, directa e intelectual al trabajo, y lo han aprobado para su publicación.
Financiación
Este trabajo ha sido financiado por el Departamento Administrativo de Ciencia, Tecnología e Innovación-COLCIENCIAS Subvención nº 123171249669 a JR-S.
Declaración de conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de cualquier relación comercial o financiera que pudiera interpretarse como un potencial conflicto de intereses.
Brown, A. L., y Mccarthy, R. E. (1974). Relación entre los virus del sarampión y del moquillo canino determinada por reacciones de hipersensibilidad de tipo retardado en perros. Nature 248, 344-345. doi: 10.1038/248344a0
PubMed Abstract | CrossRef Full Text | Google Scholar