Lipofilicitet är möjligen den viktigaste fysikalisk-kemiska egenskapen hos ett potentiellt läkemedel.Den spelar en roll för löslighet, absorption, membranpenetration, plasmaproteinbindning, distribution, CNS-påverkan och fördelning till andra vävnader eller organ, t.ex. levern, och den har en inverkan på clearancevägarna. Den är viktig för ligandigenkänning, inte bara för målproteinet utan även för CYP450-interaktioner, HERG-bindning och PXR-medierad enzyminduktion.
LogP är en komponent i Lipinskis 5-regel, en tumregel för att förutsäga löslighet och permeabilitet som har blivit ett surrogat för läkemedelsliknande egenskaper.
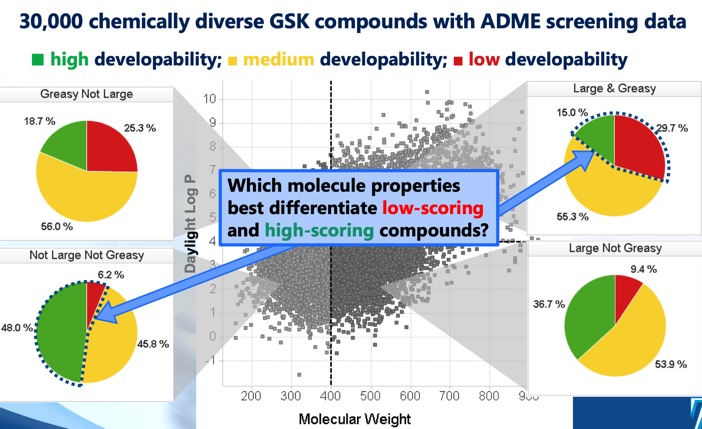
The Developability score DOI identifierar fyra distinkta cLog P/molekylvikt-regioner som definierar optimalt och suboptimalt kemiskt utrymme, och en utvecklingsbarhetspoäng som härrör från regressionsmodeller med hjälp av data från screening av löslighet, permeabilitet, proteinbindning och 3A4-hämning. Medan sektorn MWt <400, cLogP <4 föreslog den största chansen till framgång, noterades att även sektorn MWt >400, cLogP >4 innehöll en del utvecklingsbara molekyler, om än med mycket lägre chans till framgång.
Det vanligaste måttet på lipofilicitet är LogP, detta är fördelningskoefficienten för en molekyl mellan en vattenhaltig och en lipofil fas, vanligtvis oktanol och vatten.
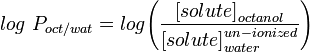
Mätning av LogP kan göras på flera olika sätt, det vanligaste är shake-flask-metoden, som går ut på att lösa upp en del av den aktuella lösta substansen i en volym oktanol och vatten, skaka under en tid och sedan mäta koncentrationen av den lösta substansen i varje lösningsmedel. Detta kan vara tidskrävande, särskilt om det inte finns någon snabb spektroskopisk metod för att mäta koncentrationen av molekylen i faserna. En snabbare metod för att bestämma log P är högpresterande vätskekromatografi. Log P för en lösta substans kan bestämmas genom att korrelera dess retentionstid med liknande föreningar med känt log P-värde doi.
Beräkning av lipofilicitet
I vanliga fall är det inte praktiskt möjligt att experimentellt bestämma log P för varje förening som tillverkas (och det kan vara av intresse att beräkna log P före syntesen) och därför används beräknade resultat, det finns ett antal programvaruverktyg som finns tillgängliga både på skrivbordet och online (använd inte för konfidentiella föreningar).
Många av dessa program fungerar genom att använda ett stort träningsdataset med kända värden för att bestämma fragmentbidrag för understrukturer och funktionella grupper, men logP är inte en enkel additiv egenskap och korrigeringstermer behövs för att ta hänsyn till närhetseffekter, H-bindning, elektroniska effekter etc., vilket framgår av nedanstående exempel.
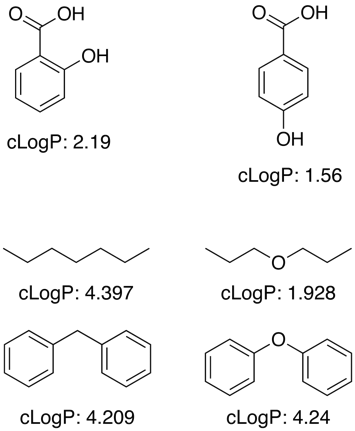
För okända funktionella grupper gör programmen ofta en approximation med hjälp av individuella atombidrag.
De olika metoderna för att beräkna logP kan delas in i tre olika tillvägagångssätt.
Atomära (t.ex. ”AlogP”, ) & Förbättrad atomisk/hybrid (”XlogP”, ”SlogP”)
Fragment (”ClogP”, KlogP, ACD/logP)
Egenskapsbaserade metoder (”MlogP”, ”VlogP”, ”MClogP”, ”TlogP”)
Atomic logP tar hänsyn till att varje atom har ett bidrag till logP och att bidragen till slutvärdet är rent additiva. Det är dock tydligt att ett kväve i en amid skiljer sig från ett kväve i en amin eller pyridin, Enhanced Atomic tar hänsyn till atomtypen.
Fragmentmetoder använder en stor träningsdatamängd med kända värden för att bestämma fragmentbidragen för understrukturer och funktionella grupper, tillsammans med korrigeringstermer för att ta hänsyn till närhetseffekter. Dessa metoder faller ofta tillbaka på atomära modeller för nya funktionella grupper.
Funktionella metoder tenderar att vara beräkningskrävande och lämpar sig inte riktigt för testning av stora datamängder.
Då träningsuppsättningarna och algoritmerna varierar mellan olika tillämpningar är det mycket viktigt att inte kombinera beräknade resultat med hjälp av olika verktyg.
En del av verktygen gör det möjligt för användaren att utöka träningsuppsättningen med hjälp av egna uppmätta värden, vilket kan vara kritiskt när man utforskar nya funktionella grupper eller ställningar.
LogD
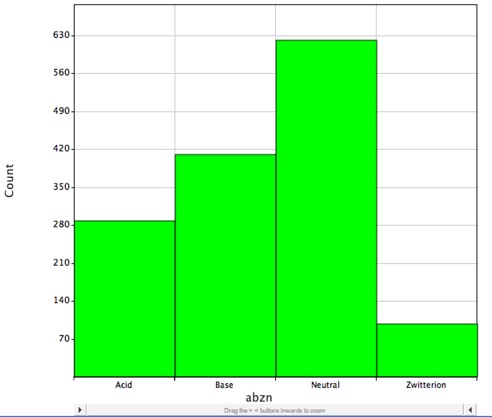
Men majoriteten av de kända läkemedlen innehåller joniserbara grupper, vilket framgår av histogrammet nedan, som visar fördelningen av småmolekylära läkemedel med DrugBank och som troligen är laddade vid fysiologiskt pH, och LogP beskriver endast fördelningskoefficienten för neutrala (oladdade) molekyler på rätt sätt.
LogD fördelningskonstanten är en bättre beskrivning av en molekyls lipofilitet. Denna kan bestämmas på liknande sätt som LogP, men i stället för att använda vatten justeras den vattenhaltiga fasen till ett visst pH med hjälp av en buffert. Log D är således pH-beroende, varför man måste ange det pH vid vilket log D mättes. Av särskilt intresse är log D vid pH = 7,4 (blodserums fysiologiska pH).
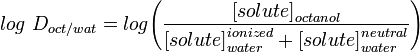
Användningar som Marvin gör det möjligt för användaren att beräkna log D men också visa pH-fördelningsprofilen, vilket visas nedan för Warfarin.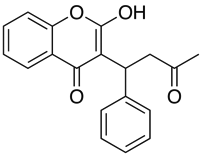
För föreningar med en pKa som ligger nära det fysiologiska pH-värdet kan det vara kritiskt att ta hänsyn till vad som faktiskt kan vara den dominerande joniserade formen.
Detta kan också vara värdefullt när man tänker på absorption från de olika regionerna i matsmältningskanalen där pH varierar från 1-3 i magsäcken till 7-8 i ileum.
De olika funktionella gruppernas bidrag till LogD har undersökts ”LogD contributions of substituents commonly used in medicinal chemistry” DOI, denna studie använde matchade molekylära paranalyser av experimentella LogD-värden från flera tusen föreningar som samlats in med hjälp av skak-flaskmetoden vid pH = 7,4. De rapporterade den genomsnittliga deltaLogD-skillnaden för särskilda molekylpar och resultaten visas nedan för det fall där den funktionella gruppen befinner sig i vilken position som helst på fenylringen. Jag har också inkluderat den beräknade LogD med hjälp av Chemaxon-programvaran.
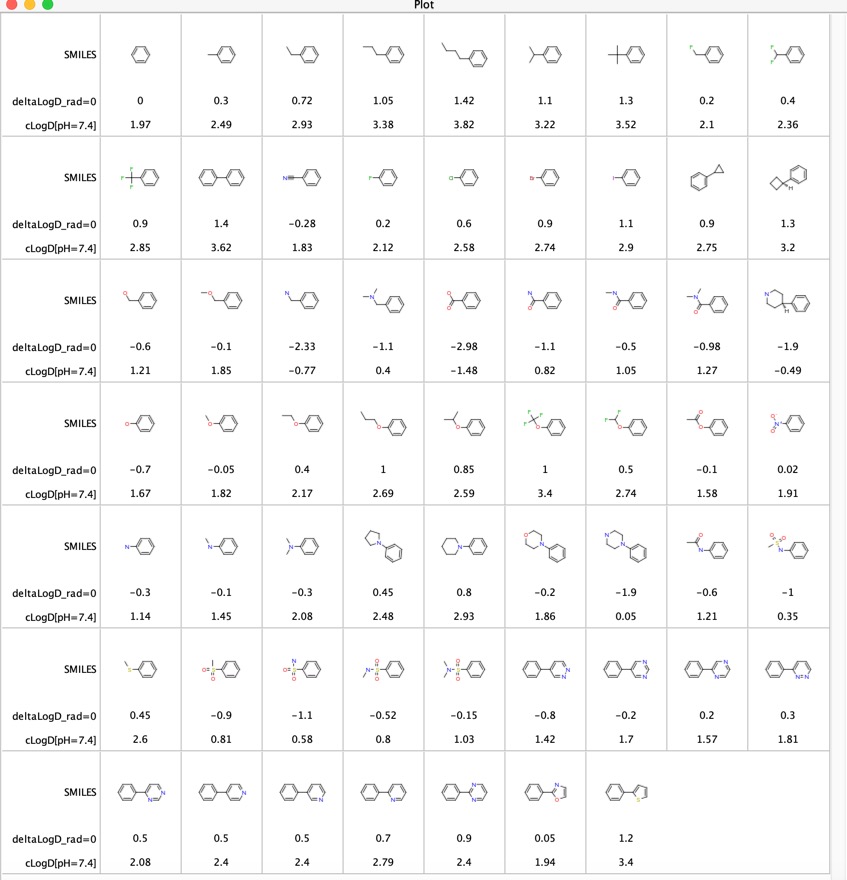
Detta är en användbar tabell för att jämföra funktionella grupper, särskilt de sista 11 posterna jämför den inverkan som olika heterocyklar har på LogD. Dessa heterocyklar används ofta som bioisosteriska ersättare för en fenylring.
Jag tänkte att det kunde vara intressant att jämföra de LogD-skillnader som bestämts med hjälp av de matchade molekylära paren (deltaLogD_rad=0) med de värden som bestämts med hjälp av den av Chemaxon beräknade LogD (deltacLogD). Som ni kan se nedan finns det en ganska bra överensstämmelse.
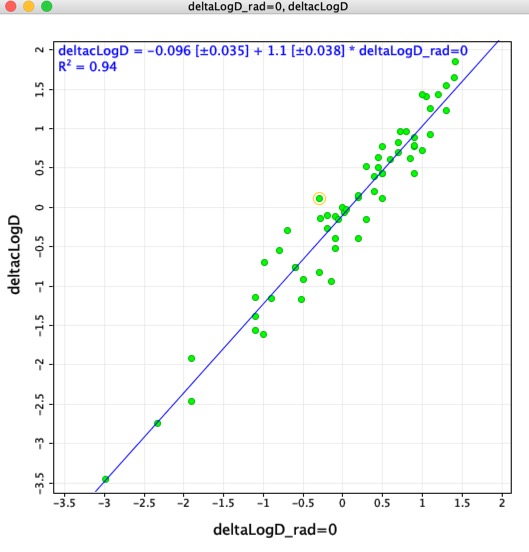
Håll dig i minnet Eftersom träningsuppsättningarna och algoritmerna varierar mellan olika tillämpningar är det mycket viktigt att inte kombinera beräknade resultat med hjälp av olika verktyg.
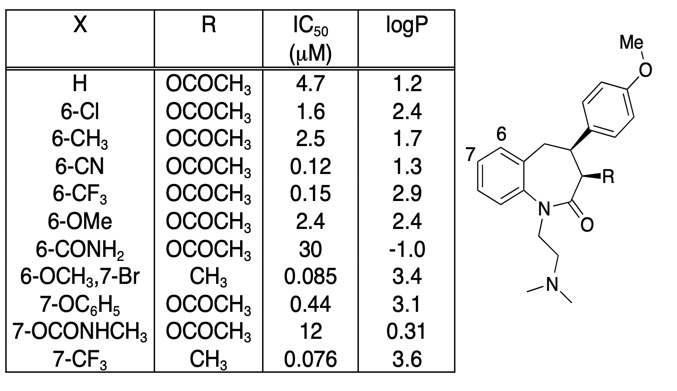
Det är viktigt att vara omdömesgill att en eventuell förbättring av bindningsaffiniteten inte helt och hållet drivs av en ökning av LogD, det är ofta användbart att helt enkelt plotta bindningsaffiniteten mot LogD. De mer intressanta sammansättningsmodifieringarna är inte nödvändigtvis de som ger den största ökningen av affiniteten utan kan vara de som ger en ökad affinitet utan motsvarande ökning av lipofiliciteten. Om man tittar på tabellen nedan finns det ett antal föreningar med mycket hög affinitet.
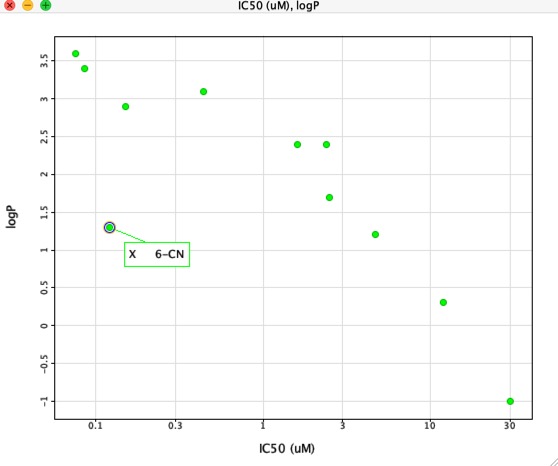
Om vi däremot plottar IC50 mot LogP som visas nedan finns det en mycket tydlig korrelation mellan LogP och IC50, men en förening skiljer sig tydligt från andra. Substituenten 6-CN ger en ökad affinitet utan motsvarande ökning av LogP.
Lipofilicitet är också en viktig komponent många av de negativa egenskaperna, inklusive plasmaproteinbindning (särskilt albumin), HERG, CYP-interaktioner, transportörer, har starka korrelationer med lipofilicitet, och det finns ett antal studier som kopplar ett högt logP till sannolikheten för att substanser misslyckas i utvecklingen på grund av dåliga ADMET-egenskaper (absorption, distribution, metabolism, utsöndring och toxicitet). Däremot är det ofta tydligt att det krävs en viss storlek och lipofilitet för att uppnå rimliga affinitetsnivåer. Att balansera dessa krav är en viktig utmaning vid upptäckt av läkemedel, och förslaget är att kemisterna bör inrikta sig på den bästa punkten, dvs. MWt 250-500 och LogP 2-4. En konsekvens av detta tillvägagångssätt är att man måste prioritera lågmolekylära, mindre lipofila föreningar vid screening. Medicinsk kemi bör inledningsvis fokusera på att välja utgångspunkter av god kvalitet och sedan kontrollera förskjutningar i fysikalisk-kemiska egenskaper på ett effektivt sätt under optimeringsprocessen.
Värt att läsa
Finding the sweet spot: the role of nature and nurture in medicinal chemistry DOI
Lipofilic efficiency: the most important efficiency metric in medicinal chemistry DOI
Sist uppdaterad 12 januari 2019
.