Morbillivirus är mycket smittsamma patogener och är ansvariga för olika utbrott i oexponerade populationer (Pfeffermann et al., 2018). De tillhör ordningen Mononegavirales och familjen Paramyxoviridae och kännetecknas av ett icke-segmenterat, linjärt, linjärt, negativt strängat RNA-genom (Lamb och Parks, 2013). Morbillivirus utmärker sig för att de orsakar måttliga till allvarliga respiratoriska, gastrointestinala, immunsupprimerande och/eller neurologiska sjukdomar hos ett stort antal värdar, inklusive människor (mässlingsvirus), köttätande djur (morbillivirus för hund, tidigare valpsjukevirus för hund), nötkreatur (rinderpestvirus), delfiner och tumlare och andra arter som är hotade av vilda djur (Lamb och Parks, 2013; Martinez-Gutierrez och Ruiz-Saenz, 2016).
Measlesvirus (MeV) och canine morbillivirus (CDV) anses vara de mest smittsamma virusen inom denna familj (De Vries et al, 2015), och på grund av CDV:s höga överföringspotential samt dess potential för överföring över artgränserna är de globala hälso- och naturvårdsmyndigheterna mycket oroade över CDV:s roll för bevarandet av utrotningshotade arter och det eventuella ”hoppet” från djur till människor (Terio och Craft, 2013; Ohishi et al., 2014). Tamhundar är huvudvärdar för CDV och kan också betraktas som en reservoar för andra däggdjur (Suzuki et al., 2015; Duque-valencia et al., 2019), men baserat på CDV:s biologi kan människor också förvandlas till en potentiell måltavla (Cosby och Weir, 2018; Rendon-Marin et al, 2019).
För att försöka förstå den potentiella risken för överföring av CDV till människor är det nödvändigt att samla alla befintliga bevis; och studiet av detta agens ursprung och spridning i hundpopulationen skulle kunna utgöra en viktig nyckel för att förstå denna process. Nyligen publicerades en artikel i International Journal of Paleopathology som inbjöd till en diskussion om CDV:s evolutionära ursprung. Slutsatsen är att CDV uppstod som en pandemisk patogen i Sydamerika efter infektion och anpassning av MeV till hundar under den sydamerikanska kolonisationsperioden. Detta resultat uppnåddes genom ett tvärvetenskapligt tillvägagångssätt som antogs genom att syntetisera en paleopatologisk analys av 96 prekolumbianska hundar (750-1470 e.Kr.) från platsen Weyanoke Old Town, Virginia, med historiska rapporter, molekylär analys och morbilliviral epidemiologi (Uhl et al, 2019).
Noterbart är att inhemska hundpopulationer från Amerika nästan försvann efter kolonisationsperioden, och europeiska och eurasiska hundar introducerades till kontinenten, vilket lämnade lite genetisk bakgrund från dess amerikanska föregångare (Ni Leathlobhair et al., 2018). En annan viktig faktor som är värd att beakta är att ”okända” sjukdomar också kan ha introducerats, vilket gör det svårare att spåra nya patogeners ursprung. Dessutom kan ett artificiellt selektionstryck på tamhundar och till och med människopopulationer, särskilt under kolonisationsperioden, ha ökat sjukdomsförekomsten och därmed begränsat den genetiska variationen (Ostrander et al, 2017), vilket i sin tur skulle kunna innebära ett mindre effektivt svar mot patogener.
Av dessa ”nya” patogener/sjukdomar beskrevs CDV först av Antonio de Ulloa y de la Torre-Giral 1746 som en sjukdom som drabbade hundar i Quito-regionen och andra delar av Sydamerika, och den rapporterades kort därefter i Europa. CDV registrerades i Spanien 1760, med 900 dödsfall under en enda dag i Madrid, och tre år senare, dvs. 1764 och 1770, hade den nått Storbritannien respektive Italien (Blancou, 2004). Virusets överförbarhet och större känslighet hos valpar jämfört med vuxna hundar rapporterades senare av Edward Jenner i början av 1800-talet. Han jämförde deras överförbarhet med MeV:s och upptäckte att överlevande var skyddade från efterföljande infektion (Jenner, 1809; Nambulli et al., 2016).
Kortfattat, efter de europeiska pionjärernas ankomst på femtonhundratalet blev nya infektionssjukdomar utan tvekan den mest förödande konsekvensen av kolonisationen, eftersom de amerikanska ursprungsbefolkningen inte hade någon tidigare exponering för patogener som hade blivit vanliga i Europa (Walker et al., 2015). Flera mässlingsepidemier ödelade därför de amerikanska ursprungsbefolkningarna (Walker et al., 2015; Nambulli et al., 2016). Uhl et al. rapporterade via ett blandat tillvägagångssätt med paleopatologiska, historiska, molekylära och epidemiologiska bevis att svåra mässlingsepidemier i de amerikanska ursprungsbefolkningarna underlättade mässlingens språng till stora tamhundspopulationer i stadsmiljöer i Sydamerika och anpassningen av viruset som endemiskt CDV (Uhl et al., 2019). Historiska uppgifter kan också bevisa att några år efter denna anpassning till sydamerikanska hundar transporterades CDV till Europa 1760, där det inledningsvis framkallade omfattande epidemier med hög dödlighet innan det blev endemiskt (Jenner, 1809).
Hur som helst beräknades molekylär fylogeografi relaterad till evolutionära förutsägelser och tiden till den senaste gemensamma förfadern (tMRCA) för CDV:s ursprung i USA på 1880-talet (95 % högsta efterföljande täthet, 1858-1913) (Panzera et al, 2015), vilket tydligt motsäger beskrivningen av viruset i Europa på 1700-talet. Sekvensanalyser som ledde till denna hypotes måste granskas noggrant på grund av bias och den begränsade tillgången på sekvenser som användes i denna molekylära fylogeografiska rekonstruktion. Dessutom har många ursprungliga förfädersekvenser gått förlorade på grund av att CDV:s och andra morbillivirus’ virala RNA-genom är labilt. Dessa faktorer har gett upphov till ifrågasättande av användbarheten av nuvarande tMRCA-beräkningar för RNA-virus (Sharp och Simmonds, 2011; Nambulli et al., 2016).
Enligt Uhl et al, morbillivirus kan ha uppkommit från nötkreatur runt 376 f.Kr. på den ”gamla kontinenten” (figur 1), och domesticering av djur kan ha haft ett betydande inflytande på händelser över artgränserna, vilket troligen spårar en startpunkt för MeV:s uppkomst till cirka 900 AC (Uhl et al., 2019). I motsats till de nuvarande CDV-fylogenetiska rekonstruktionerna stöds MeV-divergensen starkt av den bayesianska fylogenetiska analysen med avslappnad klocka. Divergenstiden mellan MeV och rinderpestviruset hade visat sig ha inträffat ungefär under det elfte till tolfte århundradet (Furuse et al., 2010). Andra molekylära data, såsom förekomsten av ett nytt morbillivirus (nära besläktat med CDV och PDV) som cirkulerar i fladdermöss från Brasilien (DrMV), gör det möjligt att spekulera i att CDV och DrMV kan ha en gemensam sydamerikansk förfader (Drexler et al., 2012), och därmed indirekt stödja idén om CDV:s tidiga sydamerikanska ursprung.
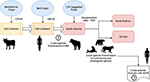
Figur 1. Schematisk representation av den möjliga evolutionära överföringsvägen för morbillivirus (CDV) hos hundar. Se texten för referenser.
Bortom den epistemologiska och/eller vetenskapliga betydelsen av det geografiska ursprunget och datumet för CDV:s divergens finns det viktiga ledtrådar som måste klarläggas för att man bättre ska kunna förstå CDV:s nuvarande påverkan på överföringen mellan arter, djurskydd och zoonotisk potential (figur 1). Det står klart att till skillnad från MeV-infektionen, som upprätthålls av en enda värd (människor), har CDV i stor utsträckning bevisats vara en promiskuös patogen som orsakar infektion/sjukdom hos ett stort antal köttätande och icke-köttätande arter (Martinez-Gutierrez och Ruiz-Saenz, 2016). Denna promiskuitet har tillskrivits inte bara CDV:s hemagglutinin (H) förmåga att interagera med värdcellsreceptorer, såsom SLAM i mononukleära celler och nectin-4 i epitelceller, utan även likheten mellan arternas sekvenser av de receptorer som nämns ovan (Rendon-Marin et al., 2019). Aminosyralikheten bland däggdjurs SLAM-receptorer, inklusive marina däggdjur, är >80 % (Ohishi et al., 2014), vilket stöder resultaten av överföring över artgränserna. Dessutom saknas artrelaterad variation i nectin-4-sekvenserna bland människor, möss och hundar eftersom mänskligt nectin-4 skulle kunna fungera som en in vitro-receptor för CDV (Noyce et al., 2011).
Naturliga CDV-utbrott hos olika icke-mänskliga primater har gett upphov till oro för en möjlig överföring av CDV till människor (Yoshikawa et al., 1989; Sun et al., 2010; Qiu et al., 2011; Sakai et al., 2013a). Det finns rapporter om att CDV-stammar från apor har den inneboende förmågan att använda mänskligt nectin-4 för att komma in i viruset och att dessa CDV från apor lätt anpassar sig till att använda den mänskliga CD150-receptorn (SLAM-receptorn) efter minimala aminosyraförändringar av virusets H-protein (Bieringer et al., 2013; Sakai et al., 2013b). Baserat på experimentell CDV-infektion in vivo av cynomolgusmakaker (Macaca fascicularis) i närvaro av MeV-immunitet var makakerna dock delvis korsskyddade från CDV-utmaningen (De Vries et al., 2014). Detta tyder på att även om CDV lätt kan infektera primater är MeV-immunitet skyddande och att CDV-infektion kan vara självbegränsande. Om man överför detta resultat till människor finns det en potentiell risk för CDV-infektion hos människor som saknar korsskyddande MeV-immunitet på grund av utebliven vaccinering och vaccinfel (Haralambieva et al., 2015) eller på grund av avsaknad av vaccinering i en eventuell era efter utrotning (Holzmann et al., 2016).
”Emerging viruses” skulle enligt uppgift kunna uppkomma via överföring av virus från djur till människor på tvärs över artgränserna (Wolfe et al., 2007). Nya studier, både strukturella och bioinformatiska, tyder på att endast en enda aminosyraförändring i en proteinsekvens kan vara tillräcklig för att övervinna begränsningen i användningen av cellulära receptorer bland två olika värdar, till exempel människor och idisslare (Abdullah et al., 2018). En unik mutation i CDV:s H-protein in vitro gör det möjligt för denna patogen att infektera celler som uttrycker den mänskliga SLAM-receptorn (Otsuki et al., 2013). Om vi antar hypotesen att CDV utvecklats från MeV skulle det dessutom vara möjligt att en CDV-avkomma skulle kunna återinfektera människor på grund av den kontinuerliga utvecklingen av både viruset och människan, vilket tidigare har föreslagits i andra modeller trots att det ursprungliga ”jumper-viruset” hade försvunnit från jorden för länge sedan (Emerman och Malik, 2010).
För övrigt är ett av de mest intressanta resultaten som presenteras av Uhl et al. optimeringen av både CDV- och MeV-gener till mänsklig kodonanvändningsbias (CUB), vilket tyder på att CDV:s kodonanvändning ligger närmare mänsklig CUB än hundarnas CUB eftersom viruset eller dess föregångare, troligen MeV, ursprungligen var anpassat till människor (Uhl et al, 2019). CUB hänvisar till fenomenet där vissa synonyma kodoner används oftare än andra och hur denna preferens varierar inom och mellan arter (Behura och Severson, 2013). I RNA-virus är kodonanvändningen under selektion eftersom virusen är helt beroende av värdarnas tRNA:er och förskjutningen beror på att virusen matchar sina värdars kodonanvändning (Jenkins och Holmes, 2003). Evolutionen kan ibland gynna virus som matchar sin värds kodonanvändning för att främja replikationshastigheten och anpassningen till värden, vilket har rapporterats i andra RNA-virus (Goni et al., 2012; Lauring et al., 2012; Di Paola et al., 2018; Freire et al., 2018).
Slutligt vill vi hävda att vissa andra faktorer måste beaktas i det möjliga zoonotiska scenariot för CDV. Korsneutralisering mellan MeV och CDV har erkänts sedan många år tillbaka (Brown och Mccarthy, 1974), och denna förutsättning har funnits i mer än ett halvt sekel när MeV-vaccinet användes för att skydda valpar mot CDV i en ålder då passiv immunitet hos modern ofta störde CDV-vaccinationen (Baker et al., 1966; Brown et al., 1972). Trots detta rekommenderas fortfarande användning av ett kommersiellt dubbelt CDV/MeV-vaccin för vaccination i närvaro av maternell immunitet, och vaccinet har varit användbart mot klinisk mässlingssjukdom hos icke-mänskliga primater (Christe et al., 2019). Därför kan man spekulera i att MeV-herdeimmunitet undviker CDV-hopp och eventuell återanpassning till människor via överföring via hundar eller vilda djur.
Slutanmärkningar
Evolutionen och ursprunget av virala patogener kan inte enkelt studeras; hädanefter är ett tvärvetenskapligt tillvägagångssätt nödvändigt för att förstå och kanske förutsäga nya möjliga virala hot mot människor. På grund av sin speciella biologi utgör virala patogener som CDV en unik modell för att förstå hopp mellan arter och zoonotisk potential hos virala agens som befinner sig mycket nära den mänskliga populationen. Förutom de traditionella molekylärfylogenetiska studierna och de paleopatologiska arbetena måste forskarna använda sig av olika metoder för att studera CDV:s ursprung och de nuvarande virus- och värdkraven för hoppning mellan arter. Införandet av beräkningsmetoder, t.ex. strukturell bioinformatik och paleovirologiska studier, skulle kunna bidra till att förutsäga och förebygga eller åtminstone ge en bättre förståelse för denna framväxande och kanske zoonotiska sjukdom ur ett annat perspektiv, där man inte bara beaktar sekvenseringsdata utan även strukturer och funktioner som nyckelinformation för detta ändamål.
Författares bidrag
Alla författare som anges har gjort ett väsentligt, direkt och intellektuellt bidrag till arbetet, och godkänt det för publicering.
Finansiering
Detta arbete fick ekonomiskt stöd från Departamento Administrativo de Ciencia, Tecnología e Innovación-COLCIENCIAS Grant No. 123171249669 till JR-S.
Intressekonfliktförklaring
Författarna förklarar att forskningen utfördes i avsaknad av kommersiella eller ekonomiska relationer som skulle kunna tolkas som en potentiell intressekonflikt.
Brown, A. L., och Mccarthy, R. E. (1974). Förhållandet mellan mässling och valpsjukevirus hos hundar fastställt genom fördröjda överkänslighetsreaktioner hos hundar. Nature 248, 344-345. doi: 10.1038/248344a0
PubMed Abstract | CrossRef Full Text | Google Scholar