La lipofilia è probabilmente la più importante proprietà fisico-chimica di un potenziale farmaco, gioca un ruolo nella solubilità, nell’assorbimento, nella penetrazione di membrana, nel legame con le proteine plasmatiche, nella distribuzione, nella penetrazione nel SNC e nel partizionamento in altri tessuti o organi come il fegato e ha un impatto sulle vie di clearance. È importante nel riconoscimento del ligando, non solo alla proteina bersaglio, ma anche nelle interazioni CYP450, nel legame HERG e nell’induzione enzimatica mediata da PXR.
Il logP è un componente della Regola di Lipinski 5, una regola empirica per prevedere la solubilità e la permeabilità che è diventata un surrogato per la somiglianza del farmaco.
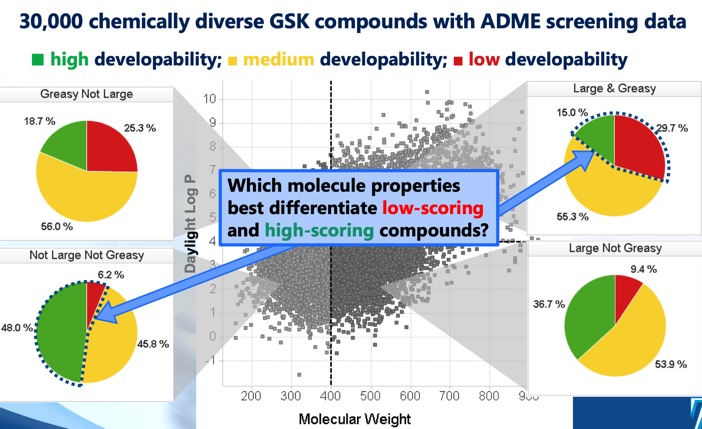
Il punteggio di sviluppabilità DOI identifica quattro distinte regioni cLog P/peso molecolare che definiscono lo spazio chimico ottimale e sub-ottimale, e un punteggio di sviluppabilità derivato da modelli di regressione utilizzando dati di screening di solubilità, permeabilità, legame proteico e inibizione 3A4. Mentre il settore MWt <400, cLogP <4 suggeriva le maggiori possibilità di successo, è stato notato che anche il settore MWt >400, cLogP >4 includeva alcune molecole sviluppabili anche se con una probabilità di successo molto più bassa.
La misura più comunemente usata della lipofilia è LogP, questo è il coefficiente di ripartizione di una molecola tra una fase acquosa e lipofila, solitamente ottanolo e acqua.
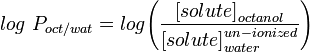
La misurazione del LogP può essere effettuata in vari modi, il più comune è il metodo dello shake-flask, che consiste nello sciogliere una parte del soluto in questione in un volume di ottanolo e acqua, agitare per un periodo di tempo, quindi misurare la concentrazione del soluto in ogni solvente. Questo può richiedere molto tempo, soprattutto se non esiste un metodo spettroscopico rapido per misurare la concentrazione della molecola nelle fasi. Un metodo più veloce per la determinazione del log P fa uso della cromatografia liquida ad alte prestazioni. Il log P di un soluto può essere determinato correlando il suo tempo di ritenzione con composti simili con valore log P noto doi.
Calcolo della lipofilia
Di solito non è pratico determinare sperimentalmente il logP di ogni composto fatto (e può essere interessante calcolare il logP prima della sintesi) e quindi si usano risultati calcolati, ci sono una serie di strumenti software disponibili sia desktop che online (non usare per composti riservati).
Molte di queste applicazioni lavorano usando un grande set di dati di addestramento di valori noti per determinare i contributi dei frammenti per le sottostrutture e i gruppi funzionali, tuttavia il logP non è una semplice proprietà additiva e sono necessari termini di correzione per permettere effetti di prossimità, legami H, effetti elettronici ecc. come mostrato negli esempi qui sotto.
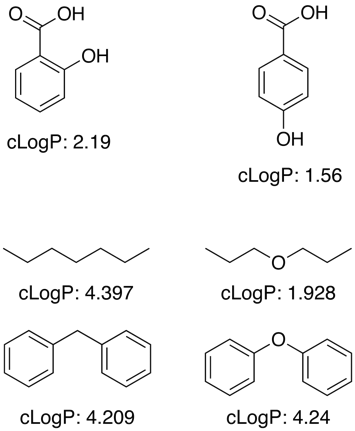
Per gruppi funzionali sconosciuti i programmi spesso approssimano usando contributi di singoli atomi.
Le diverse metodologie per calcolare il logP possono essere divise in tre diversi approcci.
Atomico (es. “AlogP”, ) &Atomico migliorato / Ibrido (“XlogP”, “SlogP”)
Frammentario (“ClogP”, KlogP, ACD/logP)
Metodi basati sulle proprietà (“MlogP”, “VlogP”, “MClogP”, “TlogP”)
Il logP atomico considera che ogni atomo ha un contributo al logP, e che i contributi al valore finale è puramente additivo. Tuttavia è chiaro che un azoto in un’ammide è diverso da un azoto in un’ammina o una piridina, Enhanced Atomic tiene conto del tipo di atomo.
I metodi per frammenti usano un grande set di dati di addestramento di valori noti per determinare i contributi dei frammenti per le sottostrutture e i gruppi funzionali, insieme a termini di correzione per tenere conto degli effetti di prossimità. Questi metodi spesso ricadono su modelli atomici per nuovi gruppi funzionali.
I metodi basati sulle proprietà tendono ad essere computazionalmente impegnativi e non sono adatti per testare grandi insiemi di dati.
Perché i set di addestramento e gli algoritmi variano tra le applicazioni, è molto importante non combinare i risultati calcolati con strumenti diversi.
Alcuni strumenti permettono all’utente di estendere l’insieme di allenamento usando valori misurati in casa, questo può essere critico quando si esplorano nuovi gruppi funzionali o scaffold.
LogD
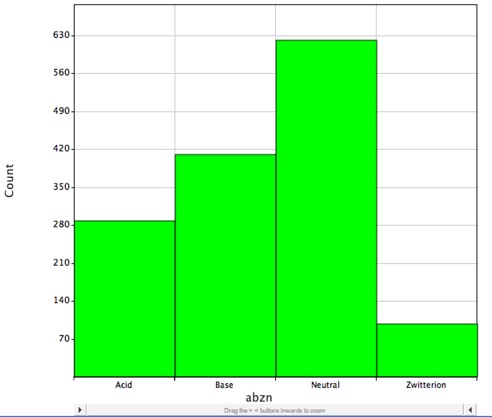
Tuttavia la maggior parte dei farmaci conosciuti contiene gruppi ionizzabili, come mostrato nell’istogramma qui sotto, questo mostra la distribuzione dei farmaci di piccole molecole con DrugBank e sono probabilmente carichi a pH fisiologico e LogP descrive correttamente solo il coefficiente di ripartizione delle molecole neutre (non cariche).
LogD la costante di distribuzione è un descrittore migliore della lipofilia di una molecola. Questo può essere determinato in modo simile al LogP, ma invece di usare l’acqua, la fase acquosa è regolata a un pH specifico usando un tampone. Il Log D è quindi dipendente dal pH, quindi si deve specificare il pH al quale il log D è stato misurato. Di particolare interesse è il log D a pH = 7,4 (il pH fisiologico del siero del sangue).
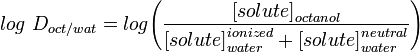
Applicazioni come Marvin permettono all’utente di calcolare il log D ma anche di visualizzare il profilo di distribuzione del pH, come mostrato sotto per il Warfarin.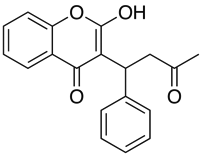
Per i composti con un pKa vicino al pH fisiologico può essere fondamentale considerare quale potrebbe essere effettivamente la forma ionizzata predominante.
Questo può anche essere prezioso quando si pensa all’assorbimento dalle diverse regioni del canale alimentare dove il pH varia da 1-3 nello stomaco a 7-8 nell’ileo.
I contributi di vari gruppi funzionali al LogD è stato esplorato “LogD contributions of substituents commonly used in medicinal chemistry” DOI, questo studio ha usato l’analisi di coppie molecolari abbinate di valori LogD sperimentali da diverse migliaia di composti raccolti utilizzando il metodo shake-flask a pH = 7.4. Hanno riportato la differenza media deltaLogD per particolari coppie molecolari e i risultati sono riportati di seguito per il caso in cui il gruppo funzionale è in qualsiasi posizione sull’anello fenilico. Ho anche incluso il LogD calcolato usando il software Chemaxon.
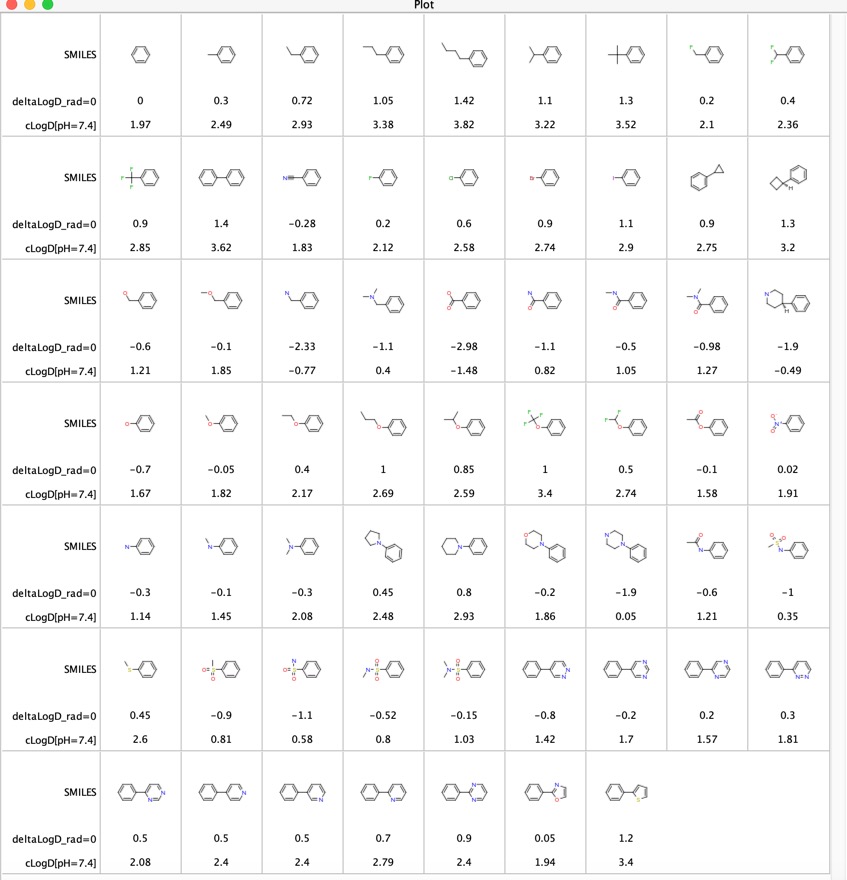
Questa è una tabella utile per confrontare i gruppi funzionali, in particolare le ultime 11 voci confrontano l’influenza di vari eterocicli hanno sul LogD. Questi eterocicli sono spesso usati come sostituzioni bioisosteriche per un anello fenile.
Ho pensato che potesse essere interessante confrontare le differenze di LogD determinate usando le coppie molecolari abbinate (deltaLogD_rad=0) con i valori determinati usando il LogD calcolato da Chemaxon (deltacLogD). Come si può vedere qui sotto c’è una corrispondenza abbastanza buona.
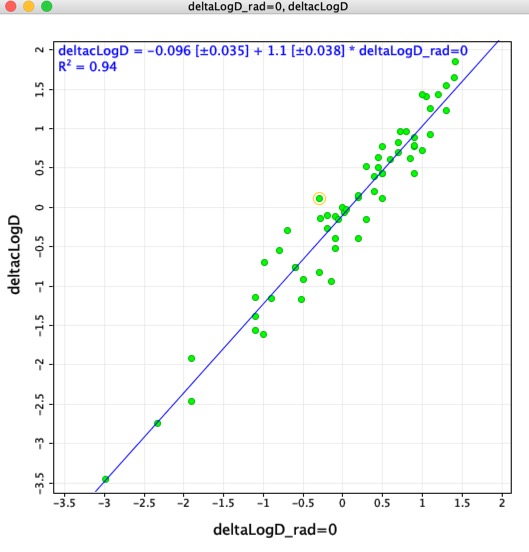
Ricorda Perché i set di allenamento e gli algoritmi variano tra le applicazioni è molto importante non combinare i risultati calcolati utilizzando strumenti diversi.
E’ importante essere cauti sul fatto che qualsiasi miglioramento dell’affinità di legame non è interamente guidato da un aumento della LogD, spesso è utile semplicemente tracciare l’affinità di legame rispetto alla LogD. Le modifiche del composto più interessanti non sono necessariamente quelle che danno il maggior aumento di affinità, ma possono essere quelle che danno un aumento di affinità senza un corrispondente aumento della lipofilia. Guardando la tabella qui sotto ci sono un certo numero di composti di affinità molto alta.
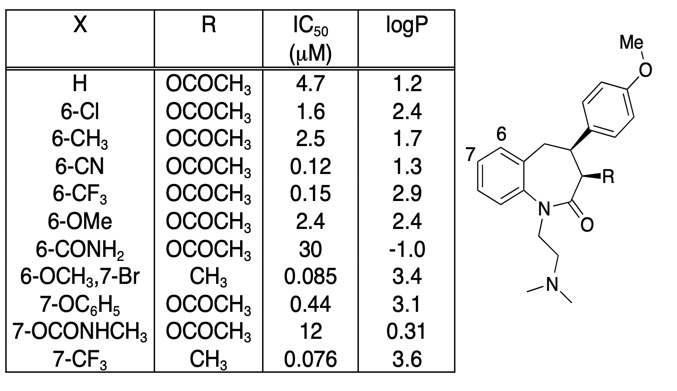
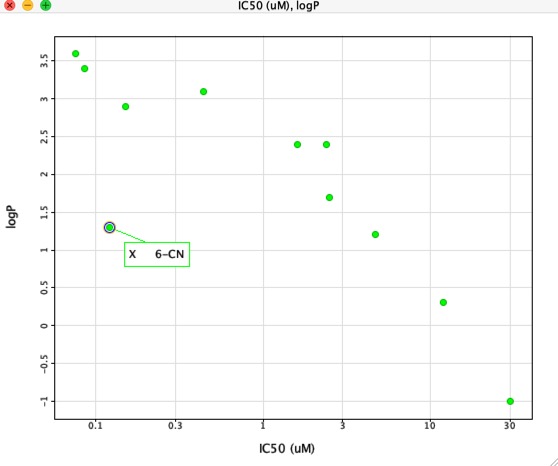
Tuttavia se tracciamo IC50 contro LogP come mostrato qui sotto c’è una correlazione molto chiara tra LogP e IC50, tuttavia un composto è chiaramente diverso. Il sostituente 6-CN dà un aumento dell’affinità senza un corrispondente aumento del LogP.
La lipofilia è anche una componente importante molte delle responsabilità off-target tra cui il legame alle proteine plasmatiche (soprattutto albumina), HERG, interazioni CYP, trasportatori, hanno forti correlazioni con la lipofilia, e ci sono stati una serie di studi che collegano un logP elevato alla probabilità che i composti falliscano nello sviluppo come risultato di scarse caratteristiche ADMET (assorbimento, distribuzione, metabolismo, escrezione e tossicità). Al contrario, è chiaro che spesso è necessaria una certa dimensione e lipofilia per ottenere livelli ragionevoli di affinità. Bilanciare questi requisiti è una sfida chiave nella scoperta della droga e il suggerimento è che i chimici mirino allo sweet spot MWt 250-500 e LogP 2-4. Una conseguenza di questo approccio è la necessità di dare la priorità ai composti a basso peso molecolare e meno lipofili dello screening. L’obiettivo iniziale della chimica medicinale dovrebbe essere quello di selezionare punti di partenza di buona qualità e poi di controllare efficacemente gli spostamenti nelle proprietà fisico-chimiche durante il processo di ottimizzazione.
Perché leggere
Finding the sweet spot: the role of nature and nurture in medicinal chemistry DOI
Lipophilic efficiency: the most important efficiency metric in medicinal chemistry DOI
Last Updated 12 January 2019