I morbillivirus sono patogeni altamente contagiosi e sono responsabili di vari focolai in popolazioni non esposte (Pfeffermann et al., 2018). Appartengono all’ordine Mononegavirales e alla famiglia Paramyxoviridae e sono caratterizzati da un genoma RNA non segmentato, lineare, a filamento negativo (Lamb e Parks, 2013). I morbillivirus si distinguono per aver causato malattie respiratorie, gastrointestinali, immunosoppressive e/o neurologiche da moderate a gravi in un’ampia gamma di ospiti, tra cui gli esseri umani (virus del morbillo), i carnivori (morbillivirus canino ex virus del cimurro canino), i bovini (virus della peste bovina), delfini e focene, e altre specie a rischio per la fauna selvatica (Lamb e Parks, 2013; Martinez-Gutierrez e Ruiz-Saenz, 2016).
Il virus del morbillo (MeV) e il morbillivirus canino (CDV) sono considerati i virus più contagiosi tra questa famiglia (De Vries et al, 2015), e a causa dell’alto potenziale di trasmissione del CDV così come il suo potenziale di trasmissione interspecie, la salute globale, e le autorità conservazioniste sono molto preoccupate per il ruolo del CDV sulla conservazione delle specie in pericolo e il possibile “salto” dagli animali agli esseri umani (Terio e Craft, 2013; Ohishi et al., 2014). I cani domestici sono l’ospite principale per il CDV e potrebbero anche essere considerati un serbatoio per altri mammiferi (Suzuki et al., 2015; Duque-valencia et al., 2019); tuttavia, sulla base della biologia del CDV, gli esseri umani potrebbero anche trasformarsi in un potenziale bersaglio (Cosby e Weir, 2018; Rendon-Marin et al, 2019).
Per cercare di comprendere il potenziale rischio di trasmissione del CDV all’uomo, è necessario raccogliere tutte le prove esistenti; e lo studio dell’origine e della diffusione di questo agente nella popolazione canina potrebbe presentare un’importante chiave di lettura di questo processo. Recentemente, un articolo pubblicato nell’International Journal of Paleopathology ha invitato ad una discussione sull’origine evolutiva del CDV. Esso conclude che il CDV ha avuto origine come patogeno pandemico in Sud America in seguito all’infezione e all’adattamento del MeV ai cani durante il periodo di colonizzazione del Sud America. Questo risultato è stato ottenuto attraverso un approccio interdisciplinare adottato sintetizzando un’analisi paleopatologica di 96 cani precolombiani (750-1470 CE) dal sito di Weyanoke Old Town, Virginia, con rapporti storici, analisi molecolare ed epidemiologia morbillivirale (Uhl et al, 2019).
In particolare, le popolazioni canine native dell’America sono quasi scomparse dopo il periodo di colonizzazione, e i cani europei ed eurasiatici sono stati introdotti nel continente, lasciando poco background genetico dei suoi predecessori americani (Ni Leathlobhair et al., 2018). Un altro fattore importante che vale la pena considerare è che potrebbero essere state introdotte anche malattie “sconosciute”, rendendo più difficile tracciare l’origine di nuovi patogeni. Inoltre, la pressione di selezione artificiale sui cani domestici e persino sulle popolazioni umane, in particolare durante il periodo di colonizzazione, potrebbe aver aumentato l’incidenza della malattia, limitando così la variazione genetica (Ostrander et al, 2017), che a sua volta potrebbe significare una risposta meno efficace contro gli agenti patogeni.
Tra questi “nuovi” patogeni/malattie, il CDV è stato descritto per la prima volta da Antonio de Ulloa y de la Torre-Giral nel 1746 come una malattia che colpisce i cani nella regione di Quito e in altre parti del Sud America, ed è stato riportato poco dopo in Europa. Il CDV fu registrato in Spagna nel 1760, con 900 morti in un solo giorno a Madrid, e 3 anni dopo, cioè nel 1764 e nel 1770, aveva raggiunto la Gran Bretagna e l’Italia, rispettivamente (Blancou, 2004). La trasmissibilità del virus e la maggiore suscettibilità dei cuccioli rispetto ai cani adulti furono poi segnalate da Edward Jenner all’inizio del 1800. Egli confrontò la loro trasmissibilità con quella del MeV e scoprì che i sopravvissuti erano protetti dall’infezione successiva (Jenner, 1809; Nambulli et al., 2016).
In breve, dopo l’arrivo dei pionieri europei nel XV secolo, le nuove malattie infettive divennero probabilmente la conseguenza più devastante della colonizzazione perché le popolazioni indigene americane non erano state precedentemente esposte agli agenti patogeni che erano diventati comuni in Europa (Walker et al., 2015). Molteplici epidemie di morbillo, quindi, devastarono le popolazioni indigene americane (Walker et al., 2015; Nambulli et al., 2016). Uhl et al. attraverso un approccio misto di prove paleopatologiche, storiche, molecolari ed epidemiologiche, hanno riferito che gravi epidemie di MeV nelle popolazioni indigene americane hanno facilitato il salto di MeV alle grandi popolazioni di cani domestici degli ambienti urbani in Sud America e l’adattamento del virus come CDV endemico (Uhl et al., 2019). Inoltre, le registrazioni storiche potrebbero dimostrare che pochi anni dopo quell’adattamento ai cani sudamericani, il CDV è stato trasportato in Europa nel 1760, dove inizialmente ha indotto epidemie diffuse con alta mortalità prima di diventare endemico (Jenner, 1809).
Tuttavia, la filogeografia molecolare relativa alle previsioni evolutive e il tempo all’antenato comune più recente (tMRCA) sono stati calcolati per l’origine del CDV negli Stati Uniti negli anni 1880 (95% massima densità posteriore, 1858-1913) (Panzera et al, 2015), che contraddice chiaramente la descrizione del virus in Europa nel XVIII secolo. Le analisi delle sequenze che hanno portato a questa ipotesi devono essere esaminate attentamente a causa dei bias e della limitata disponibilità di sequenze che sono state utilizzate in questa ricostruzione di filogeografia molecolare. Inoltre, molte sequenze ancestrali originali sono state perse a causa della labilità del genoma dell’RNA virale del CDV e di altri morbillivirus. Questi fattori hanno dato origine alla messa in discussione dell’utilità degli attuali calcoli tMRCA per i virus a RNA (Sharp e Simmonds, 2011; Nambulli et al., 2016).
Secondo Uhl et al, il morbillivirus potrebbe aver avuto origine dai bovini intorno al 376 a.C. nel “vecchio continente” (Figura 1), e la domesticazione animale potrebbe aver avuto un’influenza significativa sugli eventi cross-specie, facendo probabilmente risalire un punto di partenza nell’emergenza del MeV a circa il 900 a.C. (Uhl et al., 2019). Contrariamente alle attuali ricostruzioni filogenetiche di CDV, la divergenza di MeV è fortemente supportata dall’analisi filogenetica bayesiana dell’orologio rilassato. È stato dimostrato che il tempo di divergenza tra MeV e il virus della peste bovina è avvenuto approssimativamente tra l’undicesimo e il dodicesimo secolo (Furuse et al., 2010). Altri dati molecolari, come la presenza di un nuovo morbillivirus (strettamente correlato a CDV e PDV) che circola nei pipistrelli del Brasile (DrMV), permettono di ipotizzare che CDV e DrMV possano condividere un antenato sudamericano comune (Drexler et al., 2012), supportando così indirettamente l’idea dell’origine sudamericana precoce del CDV.
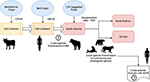
Figura 1. Rappresentazione schematica della possibile via di trasmissione evolutiva del morbillivirus canino (CDV). Vedere il testo per i riferimenti.
Al di là del significato epistemologico e/o scientifico dell’origine geografica e della data di divergenza del CDV, ci sono importanti indizi che devono essere chiariti per comprendere meglio l’attuale impatto dei CDV sulla trasmissione interspecie, la conservazione degli animali e il potenziale zoonotico (Figura 1). È chiaro che a differenza dell’infezione da MeV, che è mantenuta da un singolo ospite (gli esseri umani), CDV è stato ampiamente dimostrato di essere un patogeno promiscuo che causa infezione/malattia in una vasta gamma di specie carnivore e non carnivore (Martinez-Gutierrez e Ruiz-Saenz, 2016). Questa promiscuità è stata attribuita non solo alla capacità dell’emoagglutinina (H) del CDV di interagire con i recettori cellulari dell’ospite, come SLAM nelle cellule mononucleate e nectina-4 nelle cellule epiteliali, ma anche alla somiglianza tra le sequenze di specie dei recettori di cui sopra (Rendon-Marin et al., 2019). La somiglianza aminoacidica tra i recettori SLAM dei mammiferi, compresi i mammiferi marini, è >80% (Ohishi et al., 2014), supportando così i risultati della trasmissione cross-specie. Inoltre, c’è una mancanza di variazione specie-correlata nelle sequenze di nectina-4 tra gli esseri umani, i topi e i cani perché la nectina-4 umana potrebbe funzionare come un recettore in vitro per CDV (Noyce et al., 2011).
I focolai naturali di CDV in diversi primati non umani hanno sollevato una preoccupazione riguardo alla possibile trasmissione di CDV agli esseri umani (Yoshikawa et al., 1989; Sun et al., 2010; Qiu et al., 2011; Sakai et al., 2013a). Ci sono rapporti che i ceppi di scimmia CDV hanno la capacità intrinseca di utilizzare la nectina-4 umana per l’ingresso del virus e che questi CDV di scimmia si adattano facilmente a utilizzare il recettore CD150 umano (SLAM) in seguito a minime modifiche di aminoacidi alla proteina H virale (Bieringer et al., 2013; Sakai et al., 2013b). Tuttavia, sulla base dell’infezione sperimentale di CDV in vivo dei macachi Cynomolgus (Macaca fascicularis) in presenza di immunità MeV, i macachi erano parzialmente protetti in modo incrociato dalla sfida CDV (De Vries et al., 2014). Questo suggerisce che anche se CDV può facilmente infettare i primati, l’immunità MeV è protettiva e che l’infezione da CDV potrebbe essere autolimitante. Trasferendo questo risultato agli esseri umani, c’è un potenziale rischio di infezione da CDV nelle persone che non hanno l’immunità cross-protettiva da MeV a causa della non vaccinazione e dei fallimenti dei vaccini (Haralambieva et al., 2015) o a causa dell’assenza di vaccinazione nella possibile era post-eradicazione (Holzmann et al., 2016).
“I “virus emergenti” potrebbero sorgere tramite la trasmissione cross-specie di virus dagli animali all’uomo (Wolfe et al., 2007). Nuovi studi, sia strutturali che bioinformatici, suggeriscono che un solo cambiamento aminoacidico in una sequenza proteica potrebbe essere sufficiente per superare la restrizione nell’uso di recettori cellulari tra due ospiti diversi, come gli esseri umani e i ruminanti (Abdullah et al., 2018). Una mutazione unica nella proteina H del CDV in vitro permette a questo patogeno di infettare cellule che esprimono il recettore SLAM umano (Otsuki et al., 2013). Inoltre, se abbracciamo l’ipotesi che CDV si sia evoluto da MeV, potrebbe essere possibile che un discendente di CDV possa essere in grado di reinfettare l’uomo a causa della continua evoluzione sia del virus che dell’uomo, come è stato precedentemente suggerito in altri modelli anche se l’ancestrale “jumper virus” era scomparso dalla terra tempo fa (Emerman e Malik, 2010).
Inoltre, uno dei risultati più interessanti presentati da Uhl et al. è l’ottimizzazione di entrambi i geni CDV e MeV al codon usage bias (CUB) umano, suggerendo che l’uso del codone CDV è più vicino al CUB umano che al CUB canino perché il virus o il suo progenitore, molto probabilmente MeV, è stato inizialmente adattato agli umani (Uhl et al, 2019). CUB si riferisce al fenomeno per cui alcuni codoni sinonimi sono usati più spesso di altri e come questa preferenza varia all’interno e tra le specie (Behura e Severson, 2013). Nei virus a RNA, l’uso del codone è sotto selezione perché i virus sono completamente dipendenti dai tRNA dell’ospite e la distorsione deriva dal fatto che i virus corrispondono all’uso del codone del loro ospite (Jenkins e Holmes, 2003). L’evoluzione può talvolta favorire i virus che corrispondono all’uso del codone del loro ospite per promuovere la velocità di replicazione e l’adattamento all’ospite, come è stato riportato in altri virus a RNA (Goni et al., 2012; Lauring et al., 2012; Di Paola et al., 2018; Freire et al., 2018).
Infine, vorremmo sostenere che alcuni altri fattori devono essere considerati nel possibile scenario zoonotico del CDV. La neutralizzazione incrociata tra MeV e CDV è stata riconosciuta da molti anni (Brown e Mccarthy, 1974), e questa premessa esiste da più di mezzo secolo quando il vaccino MeV è stato utilizzato per proteggere i cuccioli contro il CDV in un’età in cui l’immunità materna passiva spesso interferiva con la vaccinazione contro il CDV (Baker et al., 1966; Brown et al., 1972). Tuttavia, l’uso di un doppio vaccino commerciale CDV/MeV è ancora raccomandato per la vaccinazione in presenza di immunità materna, e il vaccino è stato utile contro la malattia clinica del morbillo nei primati non umani (Christe et al., 2019). Quindi, si può ipotizzare che l’immunità di gregge MeV eviti il salto del CDV e il possibile riadattamento all’uomo attraverso la trasmissione attraverso cani o animali selvatici.
Raccomandazioni conclusive
L’evoluzione e l’origine dei patogeni virali non possono essere facilmente studiate; in seguito, è necessario un approccio multidisciplinare per comprendere e forse prevedere nuove possibili minacce virali per l’uomo. A causa della loro biologia peculiare, i patogeni virali come il CDV rappresentano un modello unico per comprendere il salto interspecie e il potenziale zoonotico di agenti virali molto vicini alla popolazione umana. Oltre ai tradizionali studi di filogenesi molecolare e ai lavori di paleopatologia, i ricercatori devono adottare diversi approcci per studiare l’origine del CDV e gli attuali requisiti virali e dell’ospite per il salto interspecie. L’introduzione di metodi computazionali, come la bioinformatica strutturale e gli studi di paleovirologia, potrebbe aiutare nella previsione e prevenzione o almeno fornire una migliore comprensione di questa emergente, e forse, malattia zoonotica da una prospettiva diversa, considerando non solo i dati di sequenziamento ma anche le strutture e le funzioni come informazioni chiave per questo scopo.
Contributi degli autori
Tutti gli autori elencati hanno dato un contributo sostanziale, diretto e intellettuale al lavoro e lo hanno approvato per la pubblicazione.
Finanziamento
Questo lavoro è stato sostenuto finanziariamente dal Departamento Administrativo de Ciencia, Tecnología e Innovación-COLCIENCIAS Grant No. 123171249669 a JR-S.
Dichiarazione di conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.
Brown, A. L., e Mccarthy, R. E. (1974). Relazione tra i virus del morbillo e del cimurro canino determinata da reazioni di ipersensibilità di tipo ritardato nei cani. Natura 248, 344-345. doi: 10.1038/248344a0
PubMed Abstract | CrossRef Full Text | Google Scholar