Morbilliviren sind hochansteckende Krankheitserreger und für verschiedene Ausbrüche in nicht exponierten Populationen verantwortlich (Pfeffermann et al., 2018). Sie gehören zur Ordnung der Mononegavirales und zur Familie der Paramyxoviridae und zeichnen sich durch ein nicht segmentiertes, lineares, negativ-strängiges RNA-Genom aus (Lamb und Parks, 2013). Morbilliviren zeichnen sich dadurch aus, dass sie mittelschwere bis schwere respiratorische, gastrointestinale, immunsuppressive und/oder neurologische Erkrankungen bei einer Vielzahl von Wirten verursachen, darunter Menschen (Masernvirus), Fleischfresser (canines Morbillivirus, ehemals canines Staupevirus), Rinder (Rinderpestvirus), Delfine und Schweinswale sowie andere vom Aussterben bedrohte Tierarten (Lamb und Parks, 2013; Martinez-Gutierrez und Ruiz-Saenz, 2016).
Das Masernvirus (MeV) und das canine Morbillivirus (CDV) gelten als die ansteckendsten Viren dieser Familie (De Vries et al, 2015), und aufgrund des hohen Übertragungspotenzials von CDV sowie seines artenübergreifenden Übertragungspotenzials sind die globalen Gesundheits- und Naturschutzbehörden sehr besorgt über die Rolle von CDV bei der Erhaltung gefährdeter Arten und den möglichen „Sprung“ von Tieren auf Menschen (Terio und Craft, 2013; Ohishi et al., 2014). Haushunde sind der Hauptwirt für CDV und könnten auch als Reservoir für andere Säugetiere betrachtet werden (Suzuki et al., 2015; Duque-valencia et al., 2019); aufgrund der Biologie von CDV könnte jedoch auch der Mensch zu einem potenziellen Ziel werden (Cosby und Weir, 2018; Rendon-Marin et al, 2019).
Um das potenzielle Risiko einer Übertragung von CDV auf den Menschen zu verstehen, ist es notwendig, alle vorhandenen Beweise zu sammeln; und die Untersuchung des Ursprungs und der Verbreitung dieses Erregers in der Hundepopulation könnte einen wichtigen Schlüssel zum Verständnis dieses Prozesses darstellen. Kürzlich wurde im International Journal of Paleopathology ein Artikel veröffentlicht, der zu einer Diskussion über den evolutionären Ursprung von CDV führte. Sie kommt zu dem Schluss, dass CDV als pandemischer Erreger in Südamerika entstanden ist, nachdem MeV während der Kolonialisierung Südamerikas Hunde infiziert und an sie angepasst hatte. Dieses Ergebnis wurde durch einen interdisziplinären Ansatz erzielt, der eine paläopathologische Analyse von 96 präkolumbianischen Hunden (750-1470 n. Chr.) aus dem Fundort Weyanoke Old Town, Virginia, mit historischen Berichten, molekularen Analysen und morbilliviraler Epidemiologie zusammenführte (Uhl et al, 2019).
Besonders ist anzumerken, dass die einheimischen Hundepopulationen Amerikas nach der Kolonisierungsperiode fast verschwunden sind und europäische und eurasische Hunde auf den Kontinent eingeführt wurden, so dass nur wenig genetischer Hintergrund ihrer amerikanischen Vorgänger übrig blieb (Ni Leathlobhair et al., 2018). Ein weiterer wichtiger Faktor, den es zu berücksichtigen gilt, ist, dass auch „unbekannte“ Krankheiten eingeschleppt worden sein könnten, was es schwieriger macht, den Ursprung neuer Krankheitserreger zu ermitteln. Darüber hinaus könnte der künstliche Selektionsdruck auf Haushunde und sogar menschliche Populationen, insbesondere während der Kolonisierungsphase, die Krankheitsinzidenz erhöht und damit die genetische Variation eingeschränkt haben (Ostrander et al., 2017), was wiederum eine weniger wirksame Reaktion gegen Krankheitserreger bedeuten könnte.
Unter diesen „neuen“ Erregern/Krankheiten wurde CDV erstmals von Antonio de Ulloa y de la Torre-Giral im Jahr 1746 als eine Krankheit beschrieben, die Hunde in der Region Quito und anderen Teilen Südamerikas befällt, und bald darauf wurde sie auch in Europa gemeldet. In Spanien wurde CDV 1760 mit 900 Todesfällen an einem einzigen Tag in Madrid festgestellt, und drei Jahre später, d. h. 1764 und 1770, hatte es Großbritannien bzw. Italien erreicht (Blancou, 2004). Die Übertragbarkeit des Virus und die größere Anfälligkeit von Welpen im Vergleich zu erwachsenen Hunden wurde später von Edward Jenner Anfang des 19. Jahrhunderts festgestellt. Er verglich ihre Übertragbarkeit mit der von MeV und entdeckte, dass die Überlebenden vor einer späteren Infektion geschützt waren (Jenner, 1809; Nambulli et al., 2016).
Kurz gesagt, nach der Ankunft der europäischen Pioniere im 15. Jahrhundert wurden neuartige Infektionskrankheiten wohl zur verheerendsten Folge der Kolonisierung, da die indigenen amerikanischen Bevölkerungen zuvor nicht mit Krankheitserregern in Berührung gekommen waren, die in Europa üblich geworden waren (Walker et al., 2015). Mehrere Masernepidemien verwüsteten daher die indigenen amerikanischen Bevölkerungen (Walker et al., 2015; Nambulli et al., 2016). Uhl et al. berichteten mittels eines gemischten Ansatzes aus paläopathologischen, historischen, molekularen und epidemiologischen Belegen, dass schwere Masernepidemien in den indigenen amerikanischen Populationen den Sprung der Masernepidemie auf große Haushundepopulationen in städtischen Umgebungen in Südamerika und die Anpassung des Virus als endemisches CDV erleichtert haben (Uhl et al., 2019). Außerdem konnten historische Aufzeichnungen belegen, dass CDV wenige Jahre nach dieser Anpassung an südamerikanische Hunde im Jahr 1760 nach Europa gebracht wurde, wo es zunächst weit verbreitete Epidemien mit hoher Sterblichkeit auslöste, bevor es endemisch wurde (Jenner, 1809).
Die molekulare Phylogeographie, die sich auf evolutionäre Vorhersagen und die Zeit bis zum jüngsten gemeinsamen Vorfahren (tMRCA) bezieht, wurde jedoch für den CDV-Ursprung in den Vereinigten Staaten in den 1880er Jahren berechnet (95% höchste posteriore Dichte, 1858-1913) (Panzera et al, 2015), was im klaren Widerspruch zur Beschreibung des Virus in Europa im achtzehnten Jahrhundert steht. Sequenzanalysen, die zu dieser Hypothese geführt haben, müssen aufgrund der Verzerrungen und der begrenzten Verfügbarkeit von Sequenzen, die bei dieser molekularen phylogeografischen Rekonstruktion verwendet wurden, sorgfältig geprüft werden. Außerdem sind viele ursprüngliche Vorgängersequenzen aufgrund der Labilität des viralen RNA-Genoms von CDV und anderen Morbilliviren verloren gegangen. Diese Faktoren haben dazu geführt, dass der Nutzen der aktuellen tMRCA-Berechnungen für RNA-Viren in Frage gestellt wird (Sharp und Simmonds, 2011; Nambulli et al., 2016).
Nach Uhl et al, könnte das Morbillivirus um 376 v. Chr. auf dem „alten Kontinent“ von Rindern abstammen (Abbildung 1), und die Domestizierung von Tieren könnte einen bedeutenden Einfluss auf artenübergreifende Ereignisse gehabt haben, so dass der Beginn der Entstehung von Morbillivirus wahrscheinlich auf etwa 900 n. Chr. zurückgeht (Uhl et al., 2019). Im Gegensatz zu den aktuellen phylogenetischen Rekonstruktionen von CDV wird die Divergenz von MeV durch die Bayes’sche phylogenetische Analyse mit entspannter Uhr stark unterstützt. Der Zeitpunkt der Divergenz zwischen MeV und dem Rinderpestvirus liegt nachweislich etwa im elften bis zwölften Jahrhundert (Furuse et al., 2010). Andere molekulare Daten, wie das Vorhandensein eines neuen Morbillivirus (eng verwandt mit CDV und PDV), das in Fledermäusen aus Brasilien zirkuliert (DrMV), erlauben die Spekulation, dass CDV und DrMV einen gemeinsamen südamerikanischen Vorfahren haben könnten (Drexler et al., 2012), was indirekt die Idee eines frühen südamerikanischen Ursprungs von CDV unterstützt.
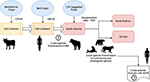
Abbildung 1. Schematische Darstellung des möglichen evolutionären Übertragungsweges des caninen Morbillivirus (CDV). Siehe Text für Referenzen.
Abgesehen von der erkenntnistheoretischen und/oder wissenschaftlichen Bedeutung des geografischen Ursprungs und des Datums der CDV-Divergenz gibt es wichtige Anhaltspunkte, die geklärt werden müssen, um die derzeitigen Auswirkungen von CDVs auf die Übertragung zwischen verschiedenen Arten, den Tierschutz und das zoonotische Potenzial besser zu verstehen (Abbildung 1). Es ist klar, dass CDV im Gegensatz zur MeV-Infektion, die von einem einzigen Wirt (dem Menschen) aufrechterhalten wird, nachweislich ein promiskuitiver Erreger ist, der Infektionen/Krankheiten bei einer Vielzahl von fleischfressenden und nicht fleischfressenden Arten verursacht (Martinez-Gutierrez und Ruiz-Saenz, 2016). Diese Promiskuität wird nicht nur auf die Fähigkeit des CDV-Hämagglutinins (H) zurückgeführt, mit zellulären Wirtsrezeptoren wie SLAM in mononukleären Zellen und Nectin-4 in Epithelzellen zu interagieren, sondern auch auf die Ähnlichkeit der Sequenzen der oben genannten Rezeptoren zwischen den Arten (Rendon-Marin et al., 2019). Die Aminosäureähnlichkeit zwischen den SLAM-Rezeptoren der Säugetiere, einschließlich der Meeressäuger, beträgt >80 % (Ohishi et al., 2014), was die Ergebnisse der artenübergreifenden Übertragung unterstützt. Darüber hinaus gibt es einen Mangel an speziesbezogener Variation in den Nectin-4-Sequenzen zwischen Menschen, Mäusen und Hunden, da menschliches Nectin-4 als In-vitro-Rezeptor für CDV fungieren könnte (Noyce et al., 2011).
Natürliche CDV-Ausbrüche bei verschiedenen nicht-menschlichen Primaten haben Bedenken hinsichtlich einer möglichen Übertragung von CDV auf den Menschen geweckt (Yoshikawa et al., 1989; Sun et al., 2010; Qiu et al., 2011; Sakai et al., 2013a). Es gibt Berichte, dass CDV-Affenstämme die intrinsische Fähigkeit besitzen, menschliches Nectin-4 für den Viruseintritt zu nutzen, und dass sich diese Affen-CDVs nach minimalen Aminosäureänderungen am viralen H-Protein leicht an den menschlichen CD150 (SLAM)-Rezeptor anpassen (Bieringer et al., 2013; Sakai et al., 2013b). Bei der experimentellen CDV-Infektion von Cynomolgus-Makaken (Macaca fascicularis) in vivo in Anwesenheit von MeV-Immunität waren die Makaken jedoch teilweise vor der CDV-Herausforderung geschützt (De Vries et al., 2014). Dies deutet darauf hin, dass CDV zwar Primaten leicht infizieren kann, die MeV-Immunität jedoch schützend wirkt und dass die CDV-Infektion selbstlimitierend sein könnte. Überträgt man dieses Ergebnis auf den Menschen, so besteht ein potenzielles Risiko einer CDV-Infektion bei Menschen, die aufgrund von Nichtimpfung und Impfversagen keine kreuzprotektive MeV-Immunität besitzen (Haralambieva et al., 2015) oder aufgrund des Fehlens einer Impfung in der möglichen Ära nach der Ausrottung (Holzmann et al., 2016).
„Emerging viruses“ könnten Berichten zufolge durch die artenübergreifende Übertragung von Viren von Tieren auf Menschen entstehen (Wolfe et al., 2007). Neue Studien, sowohl strukturelle als auch bioinformatische, deuten darauf hin, dass eine einzige Aminosäureänderung in einer Proteinsequenz ausreichen könnte, um die Beschränkung bei der Nutzung zellulärer Rezeptoren zwischen zwei verschiedenen Wirten, wie Menschen und Wiederkäuern, zu überwinden (Abdullah et al., 2018). Eine einzigartige Mutation im CDV-H-Protein in vitro ermöglicht es diesem Erreger, Zellen zu infizieren, die den menschlichen SLAM-Rezeptor exprimieren (Otsuki et al., 2013). Wenn wir uns die Hypothese zu eigen machen, dass sich CDV aus MeV entwickelt hat, könnte es außerdem möglich sein, dass ein Nachkomme von CDV in der Lage ist, den Menschen erneut zu infizieren, da sowohl das Virus als auch der Mensch eine kontinuierliche Evolution durchlaufen haben, wie dies bereits in anderen Modellen vorgeschlagen wurde, obwohl das ursprüngliche „Springervirus“ vor langer Zeit von der Erde verschwunden war (Emerman und Malik, 2010).
Eines der interessantesten Ergebnisse von Uhl et al. ist die Optimierung der CDV- und MeV-Gene auf die menschliche Codon Usage Bias (CUB), was darauf hindeutet, dass die CDV-Codon Usage näher an der menschlichen CUB als an der hündischen CUB liegt, weil das Virus oder sein Vorläufer, höchstwahrscheinlich MeV, ursprünglich an den Menschen angepasst wurde (Uhl et al., 2019). CUB bezieht sich auf das Phänomen, dass einige synonyme Codons häufiger als andere verwendet werden und dass diese Präferenz innerhalb und zwischen den Arten variiert (Behura und Severson, 2013). Bei RNA-Viren steht die Codon-Nutzung unter Selektion, da die Viren vollständig von den tRNAs ihrer Wirte abhängig sind und die Verzerrung daraus resultiert, dass die Viren die Codon-Nutzung ihrer Wirte übernehmen (Jenkins und Holmes, 2003). Die Evolution kann manchmal Viren begünstigen, die mit der Codon-Nutzung ihres Wirts übereinstimmen, um die Replikationsgeschwindigkeit und die Anpassung an den Wirt zu fördern, wie bei anderen RNA-Viren berichtet wurde (Goni et al., 2012; Lauring et al., 2012; Di Paola et al., 2018; Freire et al., 2018).
Abschließend möchten wir argumentieren, dass einige andere Faktoren in dem möglichen zoonotischen Szenario von CDV berücksichtigt werden müssen. Die Kreuzneutralisation zwischen MeV und CDV ist seit vielen Jahren bekannt (Brown und Mccarthy, 1974), und diese Prämisse besteht seit mehr als einem halben Jahrhundert, als der MeV-Impfstoff zum Schutz von Welpen gegen CDV in einem Alter verwendet wurde, in dem die passive mütterliche Immunität häufig mit der CDV-Impfung interferierte (Baker et al., 1966; Brown et al., 1972). Dennoch wird die Verwendung eines kommerziellen CDV/MeV-Doppelimpfstoffs für die Impfung bei vorhandener mütterlicher Immunität empfohlen, und der Impfstoff hat sich bei nichtmenschlichen Primaten als nützlich gegen klinische Masernerkrankungen erwiesen (Christe et al., 2019). Daher kann man spekulieren, dass die Herdenimmunität von MeV einen CDV-Sprung und eine mögliche Wiederanpassung an den Menschen über die Übertragung durch Hunde oder Wildtiere verhindert.
Abschließende Bemerkungen
Die Evolution und der Ursprung viraler Krankheitserreger lassen sich nicht ohne Weiteres erforschen; daher ist ein multidisziplinärer Ansatz erforderlich, um neue mögliche virale Bedrohungen für den Menschen zu verstehen und möglicherweise vorherzusagen. Aufgrund ihrer besonderen Biologie stellen virale Krankheitserreger wie CDV ein einzigartiges Modell für das Verständnis des Interspezies-Sprungs und des zoonotischen Potenzials von viralen Erregern dar, die der menschlichen Population sehr nahe stehen. Neben den traditionellen molekularen phylogenetischen Studien und den paläopathologischen Arbeiten müssen die Forscher verschiedene Ansätze verfolgen, um den Ursprung von CDV und die aktuellen Virus- und Wirtsanforderungen für das Springen zwischen den Arten zu untersuchen. Die Einführung computergestützter Methoden wie strukturelle Bioinformatik und paläovirologische Studien könnten bei der Vorhersage und Vorbeugung helfen oder zumindest ein besseres Verständnis dieser aufkommenden und vielleicht zoonotischen Krankheit aus einer anderen Perspektive ermöglichen, wobei nicht nur Sequenzierungsdaten, sondern auch Strukturen und Funktionen als Schlüsselinformationen für dieses Ziel berücksichtigt werden.
Autorenbeiträge
Alle aufgeführten Autoren haben einen wesentlichen, direkten und intellektuellen Beitrag zu dieser Arbeit geleistet und sie zur Veröffentlichung freigegeben.
Finanzierung
Diese Arbeit wurde finanziell unterstützt durch das Departamento Administrativo de Ciencia, Tecnología e Innovación-COLCIENCIAS Grant No. 123171249669 an JR-S.
Erklärung zu Interessenkonflikten
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Brown, A. L., and Mccarthy, R. E. (1974). Beziehung zwischen Masern- und Hundestaupeviren, bestimmt durch Überempfindlichkeitsreaktionen vom verzögerten Typ bei Hunden. Nature 248, 344-345. doi: 10.1038/248344a0
PubMed Abstract | CrossRef Full Text | Google Scholar